המומחה הרפואי של המאמר

פרסומים חדשים
מחלת שארקו-מארי-שן
סקירה אחרונה: 23.11.2021

כל תוכן iLive נבדק מבחינה רפואית או נבדק למעשה כדי להבטיח דיוק עובדתי רב ככל האפשר.
יש לנו קווים מנחים קפדניים המקור רק קישור לאתרים מדיה מכובד, מוסדות מחקר אקדמי, בכל עת אפשרי, עמיתים מבחינה רפואית מחקרים. שים לב שהמספרים בסוגריים ([1], [2] וכו ') הם קישורים הניתנים ללחיצה למחקרים אלה.
אם אתה סבור שתוכן כלשהו שלנו אינו מדויק, לא עדכני או מפוקפק אחרת, בחר אותו ולחץ על Ctrl + Enter.
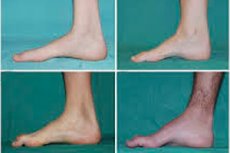
ניוון שרירים פרוניאלי, תסמונת או מחלת שארקו-מארי-שן היא קבוצה שלמה של מחלות תורשתיות כרוניות עם פגיעה בעצבים ההיקפיים.
על פי ICD-10 בסעיף המחלות של מערכת העצבים, הקוד של מחלה זו הוא G60.0 (נוירופתיה מוטורית ותורשתית תורשתית). זה נכלל גם ברשימת מחלות היתומים.
אֶפִּידֶמִיוֹלוֹגִיָה
על פי נתונים סטטיסטיים קליניים, השכיחות של כל סוגי מחלות שארקו-מארי-שן לכל 100 אלף אוכלוסיות היא 19 מקרים (על פי מקורות אחרים, מקרה אחד לכל 2.5-10 אלף אוכלוסייה).
CMT סוג 1 מהווה כשני שליש מהמקרים (מקרה אחד לכל 5-7 אלף אוכלוסייה), וכמעט 70% מהם קשורים לשכפול של הגן PMP22. בעולם, מחלה מסוג זה פוגעת ביותר מ -1.2 מיליון בני אדם.
שכיחות CMT מסוג 4 נאמדת ב 1-5 מקרים לכל 10 אלף ילדים. [1]
גורם ל מחלת שארקו-מארי-שן
על פי הסיווג של תסמונות פולי-נוירופתיות , ניוון שרירים פרונאולי (פרונאול), אמיוטרופיה עצבית של שארקו-מארי-שן או מחלת שארק-מארי-שן (בקיצור CMT) מתייחסת לפולי-נוירופתיה מוטורית-חושית. [2]
כלומר, הסיבות להופעתה הן מוטציות גנטיות. ובהתאם לאופי החריגות הגנטיות, הסוגים או הסוגים העיקריים של תסמונת זו נבדלים: demyelinating ו axonal. הקבוצה הראשונה כוללת סוג 1 של מחלת שארקו-מארי-שן (CMT1), המתרחשת כתוצאה מכפילות של הגן PMP22 בכרומוזום 17, המקודד חלבון מיאלין היקפי טרנסממברני. (תהליכים של תאי עצב) וירידה במהירות של הולכה עצבית מתרחשת. בנוסף, יתכנו מוטציות בכמה גנים אחרים.
הצורה האקסונלית היא מחלת שארקו-מארי-שן סוג 2 (CMT2), המשפיעה על האקסונים עצמם וקשורה לשינויים פתולוגיים בגן MFN2 במקום 1p36.22, המקודד לחלבון הממברנה מיטופוזין -2, הנחוץ לאיחוי מיטוכונדריאלי וליצירת רשתות מיטוכונדריאליות פונקציונליות בתוך תאי העצבים ההיקפיים. ישנם יותר מתריסר תת-סוגים של CMT2 (עם מוטציות בגנים ספציפיים).
יש לציין כי זוהו כעת יותר ממאה גנים, אשר נזקם, בירושה, גורם לתתי סוגים שונים של מחלת שארקו-מארי-שן. לדוגמא, מוטציות בגן RAB7 מפתחות CMT מסוג 2B; שינוי בגן SH3TC2 (המקודד לאחד מחלבוני קרומי תאי שוואן) גורם לסוג 4C CMT, המתבטא בילדותו ומאופיין בדמיאלינציה של נוירונים מוטוריים וחושיים (תריסר צורות וחצי מסוג 4 של מחלה זו נבדלת).
SMT מסוג 3 נדיר (הנקרא תסמונת Dejerine-Sott), הנגרם על ידי מוטציות בגנים PMP22, MPZ, EGR2 ואחרים, מתחיל להתפתח גם בגיל הרך.
כאשר CMT סוג 5 מתרחש בגיל 5-12 שנים, לא רק נוירופתיה מוטורית (בצורה של paraparesis ספסטיים של הגפיים התחתונות) נצפית, אלא גם פגיעה בעצבי הראייה והשמיעה.
חולשת שרירים וניוון עצבי הראייה (עם אובדן ראייה), כמו גם בעיות באיזון, מאפיינים סוג CMT מסוג 6. ועם מחלת שארקו-מארי-שן מסוג 7, לא נצפה רק נוירופתיה מוטורית-חושית, אלא גם מחלת רשתית בצורה של רטיניטיס פיגמנטוזה.
מחלת ה- SMT המקושרת יותר ל- XT או מחלת שארקו-מארי-שן עם טטרפרזיס של הגפיים (היחלשות התנועה של שתי הידיים והרגליים) בקרב גברים היא סוג demyelinating ונחשבת כתוצאה של מוטציה בגן GJB1 על הזרוע הארוכה של כרומוזום ה- X, המקודדת לקונקסין 32, חלבון טרנסממברני של תאי שוואן ואוליגודנדרוציטים, המסדיר את העברת אותות העצבים. [3]
גורמי סיכון
גורם הסיכון העיקרי ל- CMT הוא נוכחות של מחלה זו בהיסטוריה משפחתית, כלומר בקרובי משפחה קרובים.
לדברי גנטיקאים, אם שני ההורים הם נשאים של הגן האוטוסומאלי של מחלת שארקו-מארי-שן, הסיכון להביא לעולם ילד שיפתח מחלה זו הוא 25%. והסיכון שילד יישא את הגן הזה (אך הוא עצמו לא יסבול מסימפטומים כלשהם) נאמד בכ- 50%.
במקרה של תורשה מקושרת ל- X (כאשר הגן המוטציה נמצא על כרומוזום ה- X של האישה), קיים סיכון של 50% שהאם תעביר את הגן הזה לבנה, והוא יפתח במחלת CMT. כשנולדת ילדה, המחלה עלולה שלא להתרחש, אך בניה (נכדיה) של הבת יכולים לרשת את הגן הפגום - עם התפתחות המחלה.
פתוגנזה
בכל סוג של מחלת שארקו-מארי-שן, הפתוגנזה שלה נובעת מחריגה תורשתית של עצבים היקפיים: מוטורית (מוטורית) וחושית (חושית).
אם סוג ה- CMT ממיאלין, הריס או פגם של מעטפת המיאלין, המגן על האקסונים של העצבים ההיקפיים, מוביל להאטה בהעברת דחפים עצביים של מערכת העצבים ההיקפית - בין המוח, השרירים ואברי החישה..
בסוג האקסונלי של המחלה האקסונים מושפעים ישירות, מה שמשפיע לרעה על חוזק האותות העצביים, שאינו מספיק לגירוי מלא של שרירים ואיברי חישה.
קרא גם:
כיצד מתפשטת תסמונת שארקו-מארי-שן? גנים פגומים יכולים לעבור בירושה באופן אוטוזומלי דומיננטי או אוטוזומלי רצסיבי.
הנפוצה ביותר - תורשה דומיננטית אוטוזומלית - מתרחשת כאשר יש עותק אחד של הגן המוטציה (שנשא על ידי אחד ההורים). וההסתברות להעברת CMT לכל אחד מהילדים שנולדו נאמדת ב 50%. [4]
בתורשה אוטוזומלית רצסיבית, המחלה דורשת שני עותקים של הגן הפגום (אחד מכל הורה שאין לו סימני מחלה).
ב-40-50% מהמקרים מתרחש demyelination תורשתי דומיננטי אוטוזומלי, כלומר CMT סוג 1; ב-12-26% מהמקרים - CMT אקסונלי, כלומר סוג 2. וב 10-15% מהמקרים, נצפתה תורשה מקושרת ל- X. [5]
תסמינים מחלת שארקו-מארי-שן
בדרך כלל, הסימנים הראשונים למחלה זו מתחילים להופיע בילדות ובמתבגרים ומתפתחים בהדרגה לאורך כל החיים, אם כי התסמונת יכולה להרגיש את עצמה מאוחר יותר. שילוב הסימפטומים משתנה, ולא ניתן לחזות את קצב התקדמות המחלה, כמו גם את חומרתה.
ישנם תסמינים אופייניים בשלב הראשוני כמו עייפות כללית מוגברת; ירידה בטונוס (חולשה) של שרירי כפות הרגליים, הקרסוליים והרגליים התחתונות; חוסר רפלקסים. זה מקשה על הזזת כף הרגל ומוביל לדיסבסיה (הפרעה בהליכה) בצורה של הרמה גבוהה יותר של הרגליים, לעתים קרובות עם מעידות ונפילות תכופות. סימנים של מחלת שארקו-מארי-שן אצל ילד קטן עשויים להיות מוגדרים וקשיים בהליכה, יוצאי דופן לגיל, הקשורים לכף הרגל המשתלשלת דו - צדדית . עיוותים בכף הרגל אופייניים גם הם: קשת גבוהה (רגל חלולה) או רגליים שטוחות חזקות, אצבעות מעוקלות (דמויות פטיש).
במקרה של הליכה על בהונות על רקע לחץ דם בשרירים, הנוירולוג עשוי לחשוד שלילד יש CMT מסוג 4, בו ילדים בגיל ההתבגרות לא יוכלו ללכת.
עם התקדמותה ניוון שרירים וחולשה התפשטו לגפיים העליונות, מה שמקשה על מיומנויות מוטוריות עדינות ופעילויות יד רגילות. ירידה בתחושות המישוש והיכולת להרגיש חום וקור, כמו גם קהות ברגליים ובידיים, מעידים על פגיעה באקסונים של עצבי החישה.
כאשר באה לידי ביטוי מחלת שארקו-מארי-שן בילדות מסוגים 3 ו -6, יש אטקסיה רגישה (תיאום לקוי של תנועות ושיווי משקל), עוויתות שרירים ורעד, פגיעה בעצב הפנים, ניוון אופטי עם ניסטגמוס, אובדן שמיעה.
בשלבים מאוחרים יותר, ייתכנו רעידות (רעידות) בלתי נשלטות והתכווצויות שרירים תכופות; בעיות בתנועה עלולות להוביל להתפתחות כאב: שרירים, מפרקים, נוירופתים.
סיבוכים ותוצאות
למחלת שארקו-מארי-שן עלולים להיות סיבוכים ותוצאות כגון:
- נקעים ושברים תכופים יותר;
- התכווצויות הקשורות לקיצור השרירים והגידים הניתוחים;
- עקמת (עקמומיות של עמוד השדרה);
- בעיות נשימה - עם פגיעה בסיבי העצבים המעצבנים את שרירי הסרעפת:
- אובדן יכולת תנועה עצמאית.
אבחון מחלת שארקו-מארי-שן
האבחון כולל בדיקה קלינית, היסטוריה (כולל היסטוריה משפחתית), בדיקה נוירולוגית ומערכתית.
מבוצעות בדיקות לבדיקת טווחי תנועה, רגישות ורפלקסים בגידים. ניתן להעריך הולכת עצבים על ידי אבחון אינסטרומנטלי - אלקטרומיוגרפיה או אלקטרונורומיאוגרפיה . ייתכן שיהיה צורך גם באולטרסאונד או ב- MRI. [6]
אבחנה גנטית או DNA לזיהוי המוטציות הגנטיות הנפוצות ביותר הגורמות ל- CMT בדגימת דם מוגבלות, שכן בדיקות DNA אינן זמינות כיום לכל סוגי ה- CMT. לפרטים ראה - מחקר גנטי
במקרים מסוימים, ביופסיה של העצב ההיקפי (בדרך כלל הגסטרוקנמיוס) נעשית.
אבחון דיפרנציאלי
אבחנה דיפרנציאלית מתבצעת עם נוירופתיה היקפית אחרת, ניוון שרירים דושני, תסמונות מיאלופתיות ומיאסטניות, נוירופתיה סוכרתית, עם מיאלואפטיה במקרה של טרשת לרוחב מרובה ואמיוטרופית, תסמונת גווילין-בארה, טראומה של העצב הפרוניאלי וניוון דיסקו (כולל עמוד השדרה ), נזק למוח הקטן או לתלמוס, כמו גם לתופעות לוואי של כימותרפיה (כאשר מטפלים בציטוסטטים כגון וינקריסטין או פקליטקסל). [7]
למי לפנות?
יַחַס מחלת שארקו-מארי-שן
כיום הטיפול במחלה תורשתית זו מורכב מתרגילי פיזיותרפיה (שמטרתם חיזוק ומתיחת השרירים); ריפוי בעיסוק (המסייע לחולים עם חולשת שרירים בידיים); שימוש במכשירים אורטופדיים כדי להקל על ההליכה. במידת הצורך, קחו משככי כאבים או נוגדי פרכוסים. [8]
במקרים של רגליים שטוחות בולטות ניתן לבצע אוסטאוטומיה, ובמקרה של דפורמציה של העקבים, מצוין תיקון כירורגי שלהן - ארתרודזה. [9]
המחקר מתבצע הן על המרכיב הגנטי של המחלה והן על שיטות הטיפול בה. השימוש בתאי גזע, בחלק מההורמונים, לציטין או חומצה אסקורבית טרם הניב תוצאות חיוביות.
אך הודות למחקר העדכני ביותר, בעתיד הקרוב, באמת עשוי להופיע מחקר חדש בטיפול במחלת שארקו-מארי-שן. לכן, מאז שנת 2014 חברת Pharnext הצרפתית מפתחת, ומאז אמצע 2019, ניסויים קליניים בתרופה PXT3003 לטיפול ב- CMT סוג 1 במבוגרים, המדכאים את הביטוי המוגבר של הגן PMP22, משפרים את מיאלינה של עצבים היקפיים ו החלשת תסמינים עצב-שריריים.
מומחים של חברת הרפואה Sarepta Therapeutics (ארה"ב) עובדים על טיפול גנטי למחלת שארקו-מארי-שן מסוג 1. טיפול זה ישתמש בנגיף מזיק לאדנו (AAV) מהסוג Dependovirus עם גנום DNA חד-גדילי ליניארי, שיישא את הגן NTF3 לגוף, המקודד את החלבון הנוירוטרופין-3 (NT-3) הדרוש התפקוד של תאי עצב שוואן.
בסוף 2020, Helixmith יתחיל בניסויים קליניים של הטיפול הגנטי באנגנסיס (VM202) שפותח בדרום קוריאה לטיפול בתסמיני שרירים בסוג 1 CMT. [10]
מְנִיעָה
מניעת CMT יכולה להיות ייעוץ גנטי להורים לעתיד, במיוחד אם מישהו מזוג נשוי חולה במחלה זו במשפחה. עם זאת, זוהו מקרים של מוטציות בנקודת הגן דה נובו, כלומר בהעדר המחלה בהיסטוריה המשפחתית.
במהלך ההריון, דגימת וילה כוריונית (בין 10 ל -13 שבועות להריון), כמו גם ניתוח מי שפיר (בשבועות 15-18), מאפשרת לבדוק את הסבירות למחלת שארקו-מארי-שן אצל ילד שטרם נולד.
תַחֲזִית
באופן כללי, הפרוגנוזה לסוגים שונים של מחלת שארקו-מארי-שן תלויה בחומרתה הקלינית, אך בכל מקרה, המחלה מתקדמת לאט. לחולים רבים יש מוגבלות, אם כי אין בכך כדי להפחית את תוחלת החיים.