המומחה הרפואי של המאמר

פרסומים חדשים
דלקת כליות תורשתית (תסמונת אלפורט) אצל ילדים
סקירה אחרונה: 05.07.2025

כל תוכן iLive נבדק מבחינה רפואית או נבדק למעשה כדי להבטיח דיוק עובדתי רב ככל האפשר.
יש לנו קווים מנחים קפדניים המקור רק קישור לאתרים מדיה מכובד, מוסדות מחקר אקדמי, בכל עת אפשרי, עמיתים מבחינה רפואית מחקרים. שים לב שהמספרים בסוגריים ([1], [2] וכו ') הם קישורים הניתנים ללחיצה למחקרים אלה.
אם אתה סבור שתוכן כלשהו שלנו אינו מדויק, לא עדכני או מפוקפק אחרת, בחר אותו ולחץ על Ctrl + Enter.
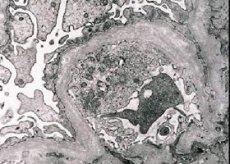
דלקת כליות תורשתית (תסמונת אלפורט) היא גלומרולופתיה תורשתית שאינה חיסונית, המתבטאת בהמטוריה (לפעמים עם חלבון בשתן), ירידה הדרגתית בתפקוד הכליות עם התפתחות אי ספיקת כליות כרונית, לעתים קרובות בשילוב עם חירשות תחושתית-עצבית וליקוי ראייה.
המחלה תוארה לראשונה בשנת 1902 על ידי ל. ג. גוטרי, שצפה במשפחה שבה נצפתה המטוריה במשך מספר דורות. בשנת 1915, א. פ. הרסט תיאר את התפתחות האורמיה אצל בני אותה משפחה. בשנת 1927, א. אלפורט זיהה לראשונה אובדן שמיעה אצל מספר קרובי משפחה עם המטוריה. בשנות ה-50 תוארו נגעים בעיניים במחלה דומה. בשנת 1972, בחולים עם המטוריה תורשתית, במהלך מחקר מורפולוגי של רקמת הכליה, הינגליס ועמיתיו חשפו התפשטות וריבוד לא אחידים של קרומי הבסיס של הגלומרולות. בשנת 1985 זוהה הבסיס הגנטי של דלקת כליות תורשתית - מוטציה בגן קולגן מסוג IV (פיינגולד ועמיתיו, 1985).
מחקר האופי הגנטי של המחלה אפשר לנו להסיק כי ההבדלים בביטויים הפנוטיפיים של דלקת כליות תורשתית (עם או בלי אובדן שמיעה) נובעים ממידת הביטוי של הגן המוטנטי. לפיכך, כיום, כל הווריאנטים הקליניים נחשבים לביטויים של מחלה אחת והמונח "דלקת כליות תורשתית" הוא שם נרדף למונח "תסמונת אלפורט".
על פי מחקרים אפידמיולוגיים, דלקת כליות תורשתית מתרחשת בשכיחות של 17 לכל 100,000 ילדים.
 [
[ גורמים לתסמונת אלפורט
הבסיס הגנטי של המחלה הוא מוטציה בגן של שרשרת a-5 של קולגן מסוג IV. סוג זה אוניברסלי עבור הקרומים הבסיסיים של הכליה, מנגנון השבלול, קפסולת העדשה, הרשתית והקרנית של העין, דבר שהוכח במחקרים המשתמשים בנוגדנים חד שבטיים כנגד מקטע קולגן זה. לאחרונה, צוינה האפשרות להשתמש בגששי DNA לאבחון טרום לידתי של דלקת כליות תורשתית.
מודגשת החשיבות של בדיקת כל בני המשפחה באמצעות בדיקות DNA לזיהוי נשאים של הגן המוטנטי, דבר בעל חשיבות רבה בייעוץ רפואי וגנטי למשפחות עם מחלה זו. עם זאת, עד 20% מהמשפחות אינן סובלות ממחלות כליות, דבר המצביע על שכיחות גבוהה של מוטציות ספונטניות של הגן החריג. לרוב החולים עם דלקת כליות תורשתית יש אנשים עם מחלת כליות, אובדן שמיעה ופתולוגיית ראייה במשפחותיהם; נישואי קרבה משפחתית בין אנשים עם אב קדמון אחד או יותר חשובים, שכן בנישואים של אנשים קרובי משפחה עולה ההסתברות לקבל את אותם גנים משני ההורים. נקבעו דרכי העברה אוטוזומליות דומיננטיות, אוטוזומליות רצסיביות ודומיננטיות, בעלות קישור X.
אצל ילדים, נבדלים לרוב שלושה סוגים של דלקת כליות תורשתית: תסמונת אלפורט, דלקת כליות תורשתית ללא אובדן שמיעה, והמטוריה שפירה משפחתית.
תסמונת אלפורט היא דלקת כליות תורשתית עם ליקוי שמיעה. היא מבוססת על פגם משולב במבנה הקולגן של קרום הבסיס הגלומרולרי של הכליות, האוזן והמבנים של העין. הגן של תסמונת אלפורט הקלאסית ממוקם בלוקוס 21-22 q של הזרוע הארוכה של כרומוזום X. ברוב המקרים, היא עוברת בתורשה באופן דומיננטי, הקשור לכרומוזום X. בהקשר זה, תסמונת אלפורט חמורה יותר אצל גברים, שכן אצל נשים תפקוד הגן המוטנטי מפוצה על ידי אלל בריא של הכרומוזום השני, שלא ניזוק.
הבסיס הגנטי להתפתחות דלקת כליות תורשתית הוא מוטציות בגנים של שרשראות אלפא של קולגן מסוג IV. ידועות שש שרשראות אלפא של קולגן מסוג IV G: הגנים של שרשראות a5 ו-a6 (Col4A5 ו-Col4A5) ממוקמים על הזרוע הארוכה של כרומוזום X באזור 21-22q; הגנים של שרשראות a3 ו-a4 (Col4A3 ו-Col4A4) נמצאים על הכרומוזום השני; הגנים של שרשראות a1 ו-a2 (Col4A1 ו-Col4A2) נמצאים על הכרומוזום ה-13.
ברוב המקרים (80-85%), מתגלה דפוס תורשה מקושר ל-X של המחלה, הקשור לנזק לגן Col4A5 כתוצאה ממחיקה, מוטציות נקודתיות או הפרעות שחבור. נכון לעכשיו, נמצאו יותר מ-200 מוטציות של הגן Col4A5, האחראיות לשיבוש הסינתזה של שרשראות a5 של קולגן מסוג IV. עם סוג זה של תורשה, המחלה מתבטאת בילדים משני המינים, אך אצל בנים היא חמורה יותר.
מוטציות בלוקוסים של הגנים Col4A3 ו-Col4A4 האחראים לסינתזה של שרשראות a3 ו-a4 של קולגן מסוג IV עוברות בתורשה אוטוזומלית. על פי מחקרים, סוג התורשה האוטוזומלי הדומיננטי נצפה ב-16% ממקרי דלקת כליות תורשתית, וסוג התורשה האוטוזומלי הרצסיבי נצפה ב-6% מהחולים. ידועים כ-10 גרסאות של מוטציות של הגנים Col4A3 ו-Col4A4.
תוצאת המוטציות היא הפרה של תהליכי ההרכבה של קולגן מסוג IV, מה שמוביל להפרה של המבנה שלו. קולגן מסוג IV הוא אחד המרכיבים העיקריים של קרום הבסיס הגלומרולרי, מנגנון השבלול ועדשת העין, אשר הפתולוגיה שלהם תתגלה במרפאה של דלקת כליות תורשתית.
קולגן מסוג IV, שהוא חלק מקרום הבסיס של הגלומרולוס, מורכב בעיקר משתי שרשראות a1 (IV) ושרשרת a2 אחת (IV), והוא מכיל גם שרשראות a3, a4 ו-a5. לרוב, בתורשה מקושרת ל-X, המוטציה של הגן Col4A5 מלווה בהיעדר שרשראות a3, a4, a5 ו-a6 במבנה של קולגן מסוג IV, ומספר שרשראות o1 ו-a2 בקרום הבסיס של הגלומרולוס גדל. המנגנון של תופעה זו אינו ברור, ההנחה היא שהסיבה היא שינויים פוסט-תעתוקיים ב-mRNA.
היעדר שרשראות a3, a4 ו-a5 במבנה הקולגן מסוג IV של קרומי הבסיס הגלומרולריים מוביל לדילולן ושבריריותן בשלבים המוקדמים של תסמונת אלפורט, המתבטאת קלינית בתדירות גבוהה יותר בהמטוריה (לפחות בהמטוריה עם פרוטאינוריה או רק פרוטאינוריה), אובדן שמיעה ולנטיקונוס. התקדמות נוספת של המחלה מובילה לעיבוי ופגיעה בחדירות של קרומי הבסיס בשלבים המאוחרים של המחלה, עם ריבוי של קולגן מסוג V ו-VI בהם, המתבטא בעלייה בפרוטאונוריה וירידה בתפקוד הכליות.
אופי המוטציה העומדת בבסיס דלקת הכליה התורשתית קובע במידה רבה את ביטויה הפנוטיפי. במקרה של מחיקה של כרומוזום X עם מוטציה בו זמנית של הגנים Col4A5 ו-Col4A6 האחראים לסינתזה של שרשראות a5 ו-a6 של קולגן מסוג IV, תסמונת אלפורט משולבת עם ליומיומטוזה של הוושט ואיברי המין. על פי נתוני מחקר, במקרה של מוטציה של הגן Col4A5 הקשורה למחיקה, נצפית חומרה גדולה יותר של התהליך הפתולוגי, שילוב של נזק כלייתי עם ביטויים חוץ-כלייתיים והתפתחות מוקדמת של אי ספיקת כליות כרונית, בהשוואה למוטציה נקודתית של גן זה.
מבחינה מורפולוגית, מיקרוסקופ אלקטרונים מגלה דילול וריבוד של קרומי הבסיס הגלומרולריים (במיוחד הלמינה דנסה) ונוכחות של גרנולות צפופות אלקטרונים. נגעים גלומרולריים עשויים להיות הטרוגניים באותו מטופל, החל מפגיעות מזנגיאליות מוקדיות מינימליות ועד גלומרולוטרשת. גלומרוליטיס בתסמונת אלפורט היא תמיד אימונו-נגטיבית, מה שמבדיל אותה מגלומרולונפריטיס. מאפיינים אופייניים כוללים התפתחות של ניוון צינורי, הסתננות לימפוהיסטיוציטית ונוכחות של "תאי קצף" עם תכלילים ליפידים - ליפופגים. ככל שהמחלה מתקדמת, מתגלים עיבוי והרס בולט של קרומי הבסיס הגלומרולריים.
מתגלים שינויים מסוימים במערכת החיסון. לחולים עם דלקת כליות תורשתית יש רמת IgA נמוכה יותר ונטייה להעלות את ריכוז ה-IgM בדם, רמת ה-IgG עשויה להיות גבוהה יותר בשלבים המוקדמים של המחלה ולרדת בשלבים המאוחרים יותר. ייתכן שהעלייה בריכוז ה-IgM וה-G היא סוג של תגובה פיצוי בתגובה לחסר ב-IgA.
הפעילות התפקודית של מערכת לימפוציטים מסוג T מצטמצמת; ישנה ירידה סלקטיבית בלימפוציטים מסוג B האחראים לסינתזה של Ig A, וקשר פגוציטי של חסינות מופרע, בעיקר עקב שיבוש כימוטקסיס ותהליכי עיכול תוך תאיים בנויטרופילים.
בבדיקת ביופסיה של כליה בחולים עם תסמונת אלפורט, נתוני מיקרוסקופ אלקטרונים מגלים שינויים אולטרה-סטרוקטורליים בקרום הבסיס הגלומרולרי: דילול, שיבוש המבנה ופיצול של קרומי הבסיס הגלומרולריים עם שינוי בעובי ובקווי המתאר הלא אחידים. בשלבים המוקדמים של דלקת כליות תורשתית, הפגם קובע את הדילול והשבירות של קרומי הבסיס הגלומרולריים.
דילול קרומי הגלומרולריות הוא סימן חיובי יותר והוא נפוץ יותר אצל בנות. סימן קבוע יותר במיקרוסקופ אלקטרונים בדלקת כליות תורשתית הוא פיצול קרום הבסיס, וחומרת הרסנו תואמת את חומרת התהליך.
תסמינים של תסמונת אלפורט אצל ילדים
התסמינים הראשונים של תסמונת אלפורט בצורת תסמונת שתן מבודדת מתגלים לרוב אצל ילדים בשלוש השנים הראשונות לחייהם. ברוב המקרים, המחלה מתגלה במקרה. תסמונת השתן מתגלה במהלך בדיקה מונעת של הילד, לפני אשפוז במוסד לטיפול בילדים או במהלך דלקת מפרקים שגרונית (ARVI). במקרה של פתולוגיה בשתן במהלך ARVI. בדלקת מפרקים שגרונית תורשתית, בניגוד לגלומרולונפריטיס נרכשת, אין תקופה סמויה.
בשלב הראשוני של המחלה, בריאותו של הילד סובלת מעט, מאפיין אופייני הוא ההתמדה והעמידות של תסמונת השתן. אחד הסימנים העיקריים הוא המטוריה בדרגות חומרה שונות, שנצפתה ב-100% מהמקרים. עלייה במידת ההמטוריה נצפית במהלך או אחרי זיהומים בדרכי הנשימה, פעילות גופנית או לאחר חיסונים מונעים. פרוטאינוריה ברוב המקרים אינה עולה על 1 גרם ליום, בתחילת המחלה יכולה להיות לא עקבית, וככל שהתהליך מתקדם, פרוטאינוריה עולה. מעת לעת, לויקוציטוריה עם דומיננטיות של לימפוציטים עשויה להופיע במשקע השתן, אשר קשורה להתפתחות שינויים ביניים.
לאחר מכן, תפקוד הכליות החלקי נפגע, מצבו הכללי של המטופל מחמיר: שכרות, חולשת שרירים, לחץ דם עורקי, לעיתים קרובות מופיעה פגיעה בשמיעה (במיוחד אצל בנים), ולעיתים פגיעה בראייה. השכבה מתבטאת בחיוורון, עייפות וכאבי ראש. בשלב הראשוני של המחלה, אובדן שמיעה מתגלה ברוב המקרים רק באמצעות אודיוגרפיה. אובדן שמיעה בתסמונת אלפורט יכול להתרחש בתקופות שונות של הילדות, אך לרוב אובדן שמיעה מאובחן בגילאי 6-10 שנים. אובדן שמיעה אצל ילדים מתחיל בתדרים גבוהים, מגיע לרמה משמעותית בהולכת אוויר ועצם, ועובר מאובדן שמיעה מוליך קול לאובדן שמיעה תופס קול. אובדן שמיעה יכול להיות אחד התסמינים הראשונים של המחלה ויכול להקדים לתסמונת השתן.
ב-20% מהמקרים, לחולים עם תסמונת אלפורט יש שינויים באיברי הראייה. האנומליות הנפוצות ביותר הן של העדשה: ספרופוקיה, לנטינוס קדמי, אחורי או מעורב, וקטרקט שונים. במשפחות עם תסמונת אלפורט, קיימת שכיחות משמעותית של קוצר ראייה. מספר חוקרים מציינים באופן קבוע שינויים דו-צדדיים פרימאקולריים במשפחות אלו בצורה של גרגירים לבנבנים או צהבהבים בהירים בגוף הצהוב. הם רואים בסימן זה סימפטום קבוע בעל ערך אבחוני גבוה בתסמונת אלפורט. KS Chugh ואחרים (1993) מצאו במחקר אופתלמולוגי בחולים עם תסמונת אלפורט ירידה בחדות הראייה ב-66.7% מהמקרים, לנטינוס קדמי ב-37.8%, כתמים ברשתית ב-22.2%, קטרקט ב-20% וקרטוקונוס ב-6.7%.
אצל חלק מהילדים הסובלים מדלקת כליות תורשתית, במיוחד כאשר מתפתחת אי ספיקת כליות, נצפה פיגור משמעותי בהתפתחות הפיזית. ככל שאי ספיקת הכליות מתקדמת, מתפתח יתר לחץ דם עורקי. אצל ילדים, הוא מזוהה לרוב בגיל ההתבגרות ובקבוצות גיל מבוגרות יותר.
חולים עם נפריטיס תורשתית מאופיינים בנוכחות של סטיגמות שונות (יותר מ-5-7) של דיסמורפוגנזה של רקמת חיבור. בין הסטיגמות של רקמת החיבור בחולים, הנפוצות ביותר הן היפרטלוריות של העיניים, חיך גבוה, אנומליות נשיכה, צורה חריגה של האופרות, עקמומיות הזרת בידיים ו"פער סנדל" בכפות הרגליים. נפריטיס תורשתית מאופיינת באחידות של סטיגמות דיסמורפוגנזה בתוך משפחה, כמו גם בתדירות גבוהה של פיזור שלהן בקרב קרובי משפחה של חולים שבשושלתם המחלה מועברת.
בשלבים המוקדמים של המחלה, מתגלה ירידה מבודדת בתפקודי הכליות החלקיים: הובלת חומצות אמינו, אלקטרוליטים, תפקוד ריכוז, חמצת, שינויים מאוחרים יותר משפיעים על המצב התפקודי של החלקים הפרוקסימליים והדיסטליים של הנפרון ומאופיינים בהפרעות חלקיות משולבות. ירידה בסינון גלומרולרי מתרחשת מאוחר יותר, לעתים קרובות יותר בגיל ההתבגרות. ככל שהנפריטיס התורשתית מתקדמת, מתפתחת אנמיה.
לפיכך, דלקת כליות תורשתית מאופיינת במהלך מדורג של המחלה: ראשית, שלב סמוי או תסמינים קליניים נסתרים, המתבטאים בשינויים מינימליים בתסמונת השתן, לאחר מכן מתרחשת פירוק הדרגתי של התהליך עם ירידה בתפקוד הכליות עם תסמינים קליניים גלויים (שיכרון, אסתניה, עיכוב התפתחותי, אנמיה). תסמינים קליניים מופיעים בדרך כלל ללא קשר לשכבות התגובה הדלקתית.
דלקת כליות תורשתית יכולה להתבטא בתקופות גיל שונות, התלויה בפעולת הגן, הנמצא במצב מודחק עד לזמן מסוים.
מִיוּן
ישנם שלושה סוגים של דלקת כליות תורשתית
- אפשרות I - מתבטאת קלינית כדלקת כליות עם המטוריה, אובדן שמיעה ונזק לעיניים. מהלך הנפריטיס הוא פרוגרסיבי עם התפתחות אי ספיקת כליות כרונית. סוג התורשה הוא דומיננטי, מקושר לכרומוזום X. מבחינה מורפולוגית, מתגלה הפרה של מבנה קרום הבסיס, דילולו ופיצולו.
- אפשרות II - מתבטאת קלינית כדלקת כליות עם המטוריה ללא אובדן שמיעה. מהלך הנפריטיס הוא פרוגרסיבי עם התפתחות אי ספיקת כליות כרונית. סוג התורשה הוא דומיננטי, מקושר לכרומוזום X. מבחינה מורפולוגית, מזוהה דילול של קרום הבסיס הנימים הגלומרולרי (במיוחד הלמינאדנסה).
- אפשרות ג' - המטוריה משפחתית שפירה. מהלך המחלה חיובי, אי ספיקת כליות כרונית אינה מתפתחת. סוג התורשה הוא אוטוזומלי דומיננטי או אוטוזומלי רצסיבי. עם סוג התורשה האוטוזומלי הרצסיבי, נצפה מהלך חמור יותר של המחלה אצל נשים.
אבחון תסמונת אלפורט
מוצעים הקריטריונים הבאים:
- נוכחות של לפחות שני חולים עם נפרופתיה בכל משפחה;
- המטוריה כתסמין המוביל של נפרופתיה אצל המטופל;
- נוכחות של אובדן שמיעה אצל לפחות בן משפחה אחד;
- התפתחות של אי ספיקת כליות כרונית אצל קרוב משפחה אחד או יותר.
באבחון מחלות תורשתיות ומולדות שונות, ניתן מקום גדול לגישה מקיפה לבדיקה, ומעל הכל, תשומת לב לנתונים המתקבלים בעת עריכת אילן היוחסין של הילד. אבחנת תסמונת אלפורט נחשבת תקפה במקרים בהם מתגלים 3 מתוך 4 סימנים אופייניים אצל המטופל: נוכחות של המטוריה ואי ספיקת כליות כרונית במשפחה, נוכחות של אובדן שמיעה נוירו-סנסורי, פתולוגיית ראייה אצל המטופל, גילוי סימני ביקוע של קרום הבסיס הגלומרולרי עם שינוי בעוביו וקווי המתאר הלא אחידים במהלך מאפיינים מיקרוסקופיים אלקטרונים של הביופסיה.
בדיקת המטופל צריכה לכלול שיטות מחקר קליניות וגנטיות; מחקר ממוקד של היסטוריית המחלה; בדיקה כללית של המטופל תוך התחשבות בקריטריונים משמעותיים מבחינה אבחנתית. בשלב הפיצוי, ניתן לאתר פתולוגיה רק על ידי התמקדות בתסמונות כגון נוכחות של נטל תורשתי, לחץ דם נמוך, סטיגמות מרובות של דיסמבריוגנזה, שינויים בתסמונת השתן. בשלב הפירוק, עשויים להופיע תסמינים חוץ-כלייתיים, כגון שכרות קשה, אסתניה, עיכוב בהתפתחות גופנית, אנמיה, המתבטאים ומתעצמים עם ירידה הדרגתית בתפקוד הכליות. ברוב החולים, עם ירידה בתפקוד הכליות, נצפים הדברים הבאים: ירידה באצידו- ואמינוגנזה; 50% מהחולים מציינים ירידה משמעותית בתפקוד ההפרשה של הכליות; טווח מוגבל של תנודות בצפיפות האופטית של השתן; הפרעה בקצב הסינון, ולאחר מכן ירידה בסינון גלומרולרי. שלב אי ספיקת כליות כרונית מאובחן כאשר לחולים יש רמה גבוהה של אוריאה בסרום הדם (מעל 0.35 גרם/ליטר) במשך 3-6 חודשים או יותר, וירידה בסינון גלומרולרי ל-25% מהנורמה.
אבחון דיפרנציאלי של דלקת כליות תורשתית צריך להתבצע בעיקר עם הצורה ההמטורית של גלומרולונפריטיס נרכשת. גלומרולונפריטיס נרכשת לרוב מופיעה באופן חריף, תקופה של 2-3 שבועות לאחר ההדבקה, סימנים חוץ-כלייתיים, כולל יתר לחץ דם מהימים הראשונים (בדלקת כליות תורשתית, לעומת זאת, לחץ דם נמוך), סינון גלומרולרי מופחת בתחילת המחלה, ללא פגיעה בתפקודים צינוריים חלקיים, בעוד שבדלקת כליות תורשתית הם קיימים. גלומרולונפריטיס נרכשת מתרחשת עם המטוריה וחלבון בשתן בולטים יותר, עם שקיעת דם מוגברת. שינויים אופייניים בקרום הבסיסי של גלומרולוס, האופייניים לנפריטיס תורשתית, הם בעלי ערך אבחוני.
אבחון מבדל של נפרופתיה דיסמטבולית מתבצע באי ספיקת כליות כרונית, במשפחה מחלות כליות הטרוגניות שזוהו קלינית, וייתכן ספקטרום של נפרופתיה מפיאלונפריטיס ועד אורוליטיאזיס. ילדים מתלוננים לעיתים קרובות על כאבים בבטן ומעת לעת במהלך מתן שתן, במשקע שתן - אוקסלטים.
אם יש חשד לדלקת כליות תורשתית, יש להפנות את המטופל למחלקה נפרולוגית מיוחדת כדי להבהיר את האבחנה.
מה צריך לבדוק?
אילו בדיקות נדרשות?
למי לפנות?
טיפול בתסמונת אלפורט
משטר הטיפול כולל הגבלות על מאמץ פיזי כבד וחשיפה לאוויר צח. התזונה מלאה, עם רמות מספיקות של חלבונים, שומנים ופחמימות מלאים, תוך התחשבות בתפקוד הכליות. חשיבות רבה היא לאיתור וטיפול במוקדי זיהום כרוניים. נעשה שימוש בתרופות הבאות: ATP, קוקורבוקסילאז, פירידוקסין (עד 50 מ"ג/יום), קרניטין כלוריד. הקורסים ניתנים 2-3 פעמים בשנה. עבור המטוריה, נקבעות תרופות צמחיות - סרפד, מיץ יער, יארו.
ישנם דיווחים בספרות זרה ומקומית על טיפול בפרדניזולון ושימוש בציטוסטטיקה. עם זאת, קשה לשפוט את ההשפעה.
באי ספיקת כליות כרונית, משתמשים בהמודיאליזה והשתלת כליה.
אין שיטות טיפול ספציפי (פתוגני יעיל) עבור דלקת כליות תורשתית. כל אמצעי הטיפול מכוונים למנוע ולהאט את הירידה בתפקוד הכליות.
התזונה צריכה להיות מאוזנת ועשירה בקלוריות, תוך התחשבות במצב התפקודי של הכליות. בהיעדר הפרעות תפקודיות, תזונת הילד צריכה להכיל מספיק חלבונים, שומנים ופחמימות. בנוכחות סימנים של תפקוד כלייתי לקוי, יש להגביל את כמות החלבון, הפחמימות, הסידן והזרחן, דבר המעכב את התפתחות אי ספיקת כליות כרונית.
יש להגביל את הפעילות הגופנית; מומלץ לילדים להימנע מספורט.
יש להימנע ממגע עם חולים זיהומיים, יש להפחית את הסיכון לפתח מחלות נשימה חריפות. יש צורך בתברואה של מוקדי זיהום כרוני. חיסונים מונעים אינם מבוצעים לילדים עם דלקת כליות תורשתית, חיסון אפשרי רק עבור אינדיקציות אפידמיולוגיות.
טיפול הורמונלי ומדכא חיסון בדלקת כליות תורשתית אינו יעיל. ישנן אינדיקציות להשפעה חיובית מסוימת (הפחתה בחלבון בשתן והאטה בהתקדמות המחלה) עם שימוש ארוך טווח ורב שנתי בציקלוספורין A ובמעכבי ACE.
בטיפול בחולים, משתמשים בתרופות המשפרות את חילוף החומרים:
- פירידוקסין - 2-3 מ"ג/ק"ג/יום ב-3 מנות למשך 4 שבועות;
- קוקארבוקסילאז - 50 מ"ג תוך שרירית כל יומיים, סה"כ 10-15 זריקות;
- ATP - 1 מ"ל תוך שרירי כל יומיים, 10-15 זריקות;
- ויטמין A - 1000 יחידות בינלאומיות לשנה ליום במנה אחת למשך שבועיים;
- ויטמין E - 1 מ"ג/ק"ג/יום במנה אחת למשך שבועיים.
טיפול מסוג זה מסייע בשיפור המצב הכללי של המטופלים, בהפחתת תפקודים לא תקינים של הצינורות ומבוצע בקורסים 3 פעמים בשנה.
ניתן להשתמש בלבמיסול כאימונומודולטור - 2 מ"ג/ק"ג/יום 2-3 פעמים בשבוע עם הפסקות בין המנות של 3-4 ימים.
על פי נתוני מחקר, לחמצון היפרברי יש השפעה חיובית על חומרת ההמטוריה ותפקוד כלייתי לקוי.
השיטה היעילה ביותר לטיפול בדלקת כליה תורשתית היא השתלת כליה בזמן. במקרה זה, אין הישנות של המחלה במהלך ההשתלה; באחוז קטן מהמקרים (כ-5%), דלקת כליה עלולה להתפתח בכליה המושתלת הקשורה לאנטיגנים לקרום הבסיסי של הגלומרול.
כיוון מבטיח הוא אבחון טרום לידתי וטיפול בהנדסה גנטית. ניסויים בבעלי חיים מראים יעילות גבוהה של העברת גנים תקינים האחראים לסינתזה של שרשראות אלפא קולגן מסוג IV לרקמת הכליה, ולאחר מכן נצפית סינתזה של מבני קולגן תקינים.
תַחֲזִית
הפרוגנוזה עבור דלקת כליות תורשתית היא תמיד חמורה.
קריטריונים פרוגנוסטיים שליליים למהלך דלקת כליות תורשתית הם:
- מין זכר;
- התפתחות מוקדמת של אי ספיקת כליות כרונית אצל בני משפחה;
- פרוטאינוריה (יותר מ-1 גרם/יום);
- עיבוי קרומי הבסיס הגלומרולריים לפי מיקרוסקופיה;
- דלקת עצב אקוסטית;
- מחיקה בגן Col4A5.
הפרוגנוזה להמטוריה משפחתית שפירה חיובית יותר.
Использованная литература