המומחה הרפואי של המאמר

פרסומים חדשים
מחלת הנטינגטון
סקירה אחרונה: 23.04.2024

כל תוכן iLive נבדק מבחינה רפואית או נבדק למעשה כדי להבטיח דיוק עובדתי רב ככל האפשר.
יש לנו קווים מנחים קפדניים המקור רק קישור לאתרים מדיה מכובד, מוסדות מחקר אקדמי, בכל עת אפשרי, עמיתים מבחינה רפואית מחקרים. שים לב שהמספרים בסוגריים ([1], [2] וכו ') הם קישורים הניתנים ללחיצה למחקרים אלה.
אם אתה סבור שתוכן כלשהו שלנו אינו מדויק, לא עדכני או מפוקפק אחרת, בחר אותו ולחץ על Ctrl + Enter.
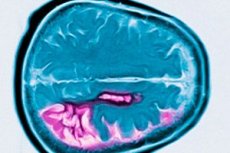
מחלת הנטינגטון היא מחלה נוירודגנרטיבית אוטוזומלית דומיננטית, המאופיינת בחולשה קוגניטיבית מתמשכת המתחילה בגיל העמידה, בתנועות לא רצוניות ובתיאום תנועות. האבחנה מאושרת על ידי בדיקה גנטית. הטיפול הוא בעיקר סימפטומטי. קרובי דם יכולים להיות מומלץ לעבור בדיקה גנטית. ג 'ורג' הנטינגטון היה הראשון לתאר את המצב הזה בשנת 1872, לאחר בחינת מקרה משפחתי של המחלה מתושבי לונג איילנד.
השכיחות של מחלת הנטינגטון היא כ -10 מקרים ל -100,000 אוכלוסייה, ובהינתן הופעתה המאוחרת, כ -30 אנשים מתוך 100,000 יש סיכון של 50% לקבל את זה בחיים שלהם. למרות שברוב המקרים המחלה מתבטאת בגיל 35-40 שנים, טווח הגילאים שלה הוא רחב למדי: ההתפרצות המוקדמת ביותר מתבטאת בגיל 3 שנים, והאחרונה - ב -90 שנים. אמנם בתחילה הוא האמין כי המחלה מאופיינת 100% החדירה, עכשיו הוא האמין כי זה לא תמיד המקרה. אצל אנשים שירשו את הגן למחלה מהאב, המחלה מופיעה בממוצע 3 שנים לפני השינה, שירשה את הגן הפתולוגי מהאם. יחד עם זאת, בכ -80% מהחולים שירשו את הגן הפתולוגי מהאב, המחלה מתבטאת עד 20 שנה. התופעה של ביטוי מוקדם יותר של פגם גנטי אצל הצאצאים נקראת ציפייה.
 [1],
[1],
מה גורם למחלת הנטינגטון?
למחלת הנטינגטון אין העדפות עדיפות. אטרופיה של גרעין caudate מוצג, שבו נוירונים קטנים להתנוון ואת רמת נוירוטרנסמיטורים - חומצה גמא aminobutyric (GABA) וחומרים פ פוחתת.
גן מוטנטי עם מספר מוגבר ("התרחבות") של רצפי דנ"א של CAG (ציסטאין-אלנין-גליצין), הקידוד של חומצת האמינו גלוטמין, אחראי על התפתחות מחלת הנטינגטון. התוצר של הגן הזה - חלבון גדול gatinging - מכיל כמות עודפת של שאריות polyglutamine, אשר מוביל למחלה על ידי מנגנון לא ידוע. ככל שחוזרת החוזרת יותר, כך המחלה מתחילה מוקדם יותר וככל שהיא מתרסקת. מדור לדור, מספר חזרות יכול להגדיל, אשר לאורך זמן מוביל להחמרה של פנוטיפ המשפחה.
למרות העניין הרב בשינויים גנטיים וביוכימיים במחלת פרקינסון, החיפוש אחר גן המחלה לא הצליח עד סוף שנות השבעים. בשלב זה, ננסי וקסלר ואלן טובין (א. טובין) ארגנו סדנה בחסות קרן תורשתית על מנת לדון באסטרטגיה לחיפוש הגן של הנטינגטון. דייויד האוסמן (ד 'האוסמן), דוד בוטשטיין (ד' ווטשטיין) וריי וייט (ר 'וייט) שהשתתפו בפגישה, הציעו כי טכניקות רקומבינציה של דנ"א שפותחו לאחרונה יכולות לסייע בהשגת מטרה זו. המשימה העיקרית בפרויקט בפיתוח היתה לחפש משפחה גדולה, שחבריה סבלו ממחלת הנטינגטון במשך דורות רבים, כדי להשיג דגימות DNA. בשנת 1979 הושק פרויקט משותף של מדענים מוונצואלה ומארה"ב, שכלל סקר של משפחה גדולה עם מחלת הנטינגטון המתגוררת בחוף אגם מאראצ'יבו (ונצואלה). בשנת 1983, המחלה של הגן הנטינגטון היה מקומי בסוף הזרוע הקצרה של כרומוזום 4 (Gusellaetal., 1983), וכן עשור מאוחר יותר התגלה כי מוטציה בגן זה היא להגדיל את מספר החזרות של trinucleotide ציטוזין-אדנין-גואנין (CAG) (הנטינגטון מחקר קבוצת מחקר שיתופית, 1993). המתודולוגיה שפותחה על ידי קבוצה מדעית זו נחשבת כיום סטנדרטית לשיבוט מיקומי של גנים חדשים.
בעוד הגן wild-type יש מתיחה של חוזרת 10-28 CAG, הצורה המוטנטית של הגן שגורמת מחלת הנטינגטון יש למתוח גדל מ 39 ליותר מ -100 חוזר CAG. זיהוי ההתרחבות של חוזר trinucleotide אפשרה לנו להסביר תכונות קליניות רבות של המחלה. בפרט, נמצא קשר הפוך בין גיל ההתפרצות לבין אורכו של האתר עם טרינוקליאוטידים חוזרים. ציפייה לירושה אבהית יכולה להיות מוסברת על ידי העובדה כי עלייה במספר החזרות מתרחשת לעתים קרובות אצל גברים במהלך spermatogenesis. ניתוח המוטציות החדשות הראה כי הן בדרך כלל מתעוררות כאשר אחד ההורים, בדרך כלל האב, היה מספר חוזר של CAG גבוה מ 28; במקרה זה, מספר החזרות גדל בדור הבא. כעת נקבע כי אם מספר החזרות הוא לא יותר מ 28, אז זה מועבר ביציבות מדור לדור. אם מספר החזרות הוא בין 29 ל -35, אז הסימפטומים של מחלת הנטינגטון אינם מופיעים, אבל כאשר מועברים לצאצאים, אורך זה עשוי לגדול. אם מספר החזרות הוא בין 36 ל -39, אז במקרים מסוימים (אך לא תמיד) המחלה עשויה להתבטא קלינית (חדירה חלקית), ועל ידי העברת לצאצאים, עלייה במספר חוזר trinucleotide עלול להתרחש. אם מספר החזרות עולה על 40, אז המחלה מתרחשת כמעט בכל המקרים, ועם העברת צאצאים, הרחבה נוספת של חזרות אפשרי. הסיבות לעלייה במספר החזרות נותרו בלתי ידועות.
פאתומורפולוגיה של מחלת הנטינגטון
מחלת הנטינגטון מאופיינת על ידי מוות של נוירונים בעיקר בגרעין caudate ואת הקליפה, במידה מסוימת גם בקליפת המוח ומבנים אחרים של המוח. המשקל הכולל של המוח במחלת הנטינגטון מצטמצם לא רק על ידי צמצום מספר הנוירונים, אלא בשל אובדן חומר לבן. בקליפת המוח, תאים בשכבות V ו- VI מושפעים ביותר. חומרת השינויים הניוונריים המיקרוסקופיים והמיקרוסקופיים (עם תיקון גיל בזמן המוות) עולה בקנה אחד עם מספר החזרת החוזר. ניתוח מפורט של שינויים בכמה מאות מקרים של מחלת הנטינגטון הראה כי התנוונות הסטריאטום מתחילה עם החלק הדורוסדיאלי של הגרעין הקוקאטי והקטע הדסורולראלי של הקליפה, ואז מתפשט בכיוון הגחון. קבוצות שונות של נוירונים של הגרעין caudate ואת הקליפה לא סובלים באותה מידה. נוירונים שהוכנסו בסטריאטום נותרו על כנם, אך נוירונים מסוימים מושפעים באופן סלקטיבי. בצורת הנער של מחלת הנטינגטון, שינויים פתולוגיים בסטריאטום בולטים יותר ויותר נפוצים, המערבים את קליפת המוח, המוח הקטן, התלמוס, הכדור החיוור.
שינויים נוירוכימיים במחלת הנטינגטון
GABA. מחקר נוירוכימי של המוח בחולים עם מחלת הנטינגטון גילה ירידה משמעותית בריכוז ה- GABA בסטריאטום. מחקרים שנערכו לאחר מכן אישרו כי מספר הנוירונים GABAergic מצטמצם מחלת הנטינגטון, והראה כי הריכוז של GABA מצטמצם לא רק בסטריאטום, אלא גם באזורי ההקרנה שלו - החלקים החיצוניים והפנימיים של כדור הארץ החיוור, כמו גם את הניגרה המהותית. במוח של מחלת הנטינגטון, השתנה גם קולטני GABA באמצעות מחייב קולטן והכלאה באתרו של ה- mRNA, מספרם של קולטני GABA צומצם במידה בינונית בגרעין ובקליפת הזנב, אך גדל בחלק הרטיקולרי של הניגרה המהותית והקטע החיצוני של כדור הארץ החיוור,, בשל רגישות יתר.
אצטילכולין. אצטילכולין משמש נוירוטרנסמיטר עבור נוירונים גדולים לא גלוי intercalary בסטריאטום. במחקר מוקדם שלאחר המוות בחולים עם מחלת הנטינגטון, זוהתה ירידה בפעילות cholinecetyltransferase (HAT) בסטריאטום, דבר שעלול להצביע על אובדן נוירונים קולינרגיים. עם זאת, בהשוואה לירידה משמעותית במספר הנוירונים GABAergic, נוירונים intercalated cholinergic להישאר שלם לחלוטין. כתוצאה מכך, הצפיפות של נוירונים חיוביים acetylcholinase ופעילות של HAT בסטריאטום הם למעשה גבוהים יחסית לעומת בקרות כי הם מאוזנים לגיל.
חומר ר 'חומר P הוא הכיל נוירונים רבים styloid בינוני של הסטריאטום, אשר מוקרנים בעיקר על החלק הפנימי של הכדור החיוור ואת מהות nigra ובדרך כלל גם מכילים dorforph ו GABA. רמת החומר P של הסטריאטום ואת החלק הרטייקולרי של הניגרה המהותית מצטמצמת במחלת הנטינגטון. בשלב המסוף של המחלה באמצעות מחקרים אימונוהיסטוכימיים חשף ירידה משמעותית במספר הנוירונים המכילים חומר R. בשלב מוקדם יותר, נוירונים המכילים חומר P ו מוקרן על החלק הפנימי של הכדור החיוור נשמרים יחסית לעומת נוירונים המקרין על החלק הרשתית של החומר השחור.
פפטידים אופיואידים. אנקפאלין נמצא בתאי הגיאגרגיים המוקרנים על-ידי הרפואה של המסלול העקיף, המוקרן על החלק החיצוני של הכדור החיוור ונושא את קולטני ה- D2 על עצמם. באמצעות מחקרים אימונוהיסטוכימיים, הוכח כי בשלב מוקדם של מחלת הנטינגטון, יש אובדן של נוירונים המכילים אנקפאלין המקרין על החלק החיצוני של הכדור החיוור. תאים אלה, ככל הנראה, למות מוקדם יותר מאשר תאים המכילים חומר P ו מקרין על החלק הפנימי של הכדור החיוור.
קטכולאמינים. נוירונים המכילים אמינים ביוגניים (דופמין, סרוטונין) ו מוקרן על הסטריאטום ממוקמים בחלק קומפקטי של ניגריה מהות, מכסה הגחון ואת תפר גרעינים. בעוד תחזיות nadrenrenergic לתוך הסטריאטום של בני אדם הם מינימליים, רמות של סרוטונין ודופאמין (במונחים של גרם של רקמה) ב הסטריאטום הם גבוהות, המציין את הבטיחות של התחזיות האלה affarent על רקע אובדן מובהק של נוירונים סטריאטליים משלהם. נוירונים דופאמינרגיים של הניגרה המהותית נשארים ללא פגע הן בצורות הקלאסיות והן אצל הצעירים של מחלת הנטינגטון.
סומאטוסטטין / נוירופפטיד Y ו- synthetase תחמוצת החנקן. מדידת רמת סומאוסטטין ו neuropeptide Y ב striatum ב הנטינגטון של המחלה גילה עלייה פי 4-5 שלהם לעומת רקמות רגילות. באמצעות מחקרים immunohistochemical, בטיחות מוחלטת של נוירונים sterstum interstitial המכיל neuropeptide Y, סומטוסטאטין ו synthetase תחמוצת החנקן נאמר. לפיכך, נוירונים אלה עמידים לתהליך הפתולוגי.
חומצות אמינו מרגשות. הוצע שמוות תאים סלקטיבי במחלת הנטינגטון קשור להשפעה נוירוטוקסית של גלוטמט. גלוטמט, ורמות חומצה קואינולינית (עצבי אנדוגניים, שהוא תוצר לוואי של חילוף החומרים של אגוניסט סרוטונין להיות glugamatnyh retsptorov) בסטריאטום של מחלת הנטינגטון עם שינוי קל, אך מחקר שנערך לאחרונה באמצעות MR - ספקטרוסקופיה חשפו את in vivo עליית גלוטמט. רמת האנזים גליה האחראי על סינתזה של חומצה quinolinic ב הסטריאטום של הנטינגטון של המחלה הוא גדל בכ 5 פעמים לעומת הנורמה, בעוד הפעילות של האנזים המספק השפלה של חומצה quinolinic הוא גדל במחלת הנטינגטון רק על ידי 20-50%. לכן, סינתזת חומצה quinolinic במחלת הנטינגטון יכול להיות משופר.
מחקרים של קולטנים של החומצות האמיניות המעוררות (HAC) במחלת הנטינגטון הראו ירידה משמעותית במספר של NMDA, AMPA, kainate ו metabotropic קולטנים glugamatnyh בסטריאטום ו AMPA ו קולטנים kainate בקליפת המוח. בשלב מאוחר של מחלת הנטינגטון, קולטני NMDA היו למעשה נעדרים, בשלב הפרה קליני ובשלבים המוקדמים הייתה ירידה משמעותית במספר הקולטנים הללו.
רגישות סלקטיבית. במחלת הנטינגטון, סוגים מסוימים של תאים סטריאטאליים מתים באופן סלקטיבי. הנוירונים הסגנוניים האמצעיים המוקרנים על החלק החיצוני של הכדור החיוור ומכילים GABA ו- enkephalin כבר מתים בשלב מוקדם מאוד של המחלה, כמו גם נוירונים המכילים GABA וחומר P ומקרבים לחלק הרטיקולרי של הניגרה. אובדן נוירונים המכילים GABA ו enkephalin ו מקרין על החלק החיצוני של הכדור חיוור מנשקה את המבנה הזה, אשר, בתורו, מוביל עיכוב פעיל של הגרעין subthalamic. את הירידה בפעילות הגרעין התת-קרקעי ניתן להסביר, ככל הנראה, על ידי התנועות הכוריאוריות המתרחשות במחלת הנטינגטון. זה כבר זמן רב ידוע כי נגעים המוקד של הגרעין subtalamic יכול להיות הגורם של chorea. איבוד נוירונים המכילים GABA וחומר P והקרנה על החלק הרטייקולרי של הניגרה המהותית יכול להיות הסיבה להפרעות אוקלומטוריות שנצפו במחלת הנטינגטון. דרך זו בדרך כלל מעכבת נוירונים של החלק הרטייקולרי של הניגרה המהותית, המוקרנת על הגבעות העליונות של מרובעת, אשר, בתורו, מווסתים saccades. בצורת הנער של מחלת הנטינגטון, השבילים שהוזכרו לעיל סובלים בצורה חמורה יותר, בנוסף, תחזיות striatal לקטע הפנימי של הכדור החיוור אבדו מוקדם.
חלבון הציד המקודד על ידי הגן, המוטציה הגורמת למחלת הנטינגטון, מזוהה במבנים שונים במוח וברקמות אחרות. בדרך כלל, huntingtin נמצא בעיקר בציטופלסמה של נוירונים. חלבון מזוהה ברוב הנוירונים של המוח, אבל, כפי שמראים הנתונים האחרונים, התוכן שלו גבוה יותר במטריצה מאשר נוירונים סטריוסומיים, ובנוירונים הקרנה גבוה יותר מאשר נוירונים intercalated. לפיכך, הרגישות הסלקטיבית של נוירונים מתואמת עם התוכן של huntingtin בהם, אשר מיוצגת בדרך כלל באוכלוסיות מסוימות של נוירונים.
כמו במוחם של מטופלים עם מחלת הנטינגטון, בעכברים מהונדסים עבור קטע N- מסוף של הגן של הנטינגטון עם מספר מוגבר של חוזר, huntingtin צורות אגרגטים צפופים בגרעינים של נוירונים. אלה תכלילים intranuclear נוצרים הקרנה steratal (אבל לא בין נוירונים). בעכברים מהונדסים, תכלילים טופס מספר שבועות לפני תחילת הסימפטומים. נתונים אלה מראים כי חלבון huntingtin, אשר מכיל מספר מוגבר של שאריות גלוטמין, הכללת אשר מקודד חוזר trinucletide, או שבר שלה מצטבר בגרעין, וכתוצאה מכך, את השליטה של פונקציות הסלולר כי הוא עלול לסבול.
סימפטומים של מחלת הנטינגטון
הגיל שבו הופיעו הסימפטומים הראשונים, בחולים עם מחלת הנטינגטון, קשה לקבוע במדויק, שכן המחלה מתבטאת בהדרגה. שינויים באישיות ובהתנהגות, הפרעות קואורדינציה קלות יכולות להתרחש שנים רבות לפני הופעתם של סימפטומים בולטים יותר. עד האבחנה, תנועות קוריאניות, תיאום לקוי של תנועות עדינות, והאטה ביצירת שרידים שרירותיים נמצאים ברוב החולים. ככל שהמחלה מתקדמת, היכולת לארגן את פעילותה נפגעת, הזיכרון מצטמצם, הדיבור נעשה קשה, ליקויים oculomotor ואת הביצועים לקוי של תנועות מתואמות להגדיל. למרות שבשלב מוקדם של המחלה אין שינויים בשרירים ובמצבים, בשל התקדמותה, תנוחות דיסטוניות יכולות להתפתח, אשר לאורך זמן יכולות להפוך לסימפטום הדומיננטי. בשלב מאוחר, הדיבור הופך להיות בלתי מובן, בליעה הופך להיות הרבה יותר קשה, הליכה הופך בלתי אפשרי. מחלת הנטינגטון מתקדמת בדרך כלל תוך 15-20 שנה. בשלב הטרמינל, החולה חסר אונים וצריך טיפול מתמיד. תוצאה קטלנית אינה קשורה ישירות למחלה העיקרית, אלא לסיבוכים שלה, למשל, דלקת ריאות.
דמנציה במחלת הנטינגטון
קוד ICD-10
Р02.2. דמנציה במחלת הנטינגטון (G10).
דמנציה מתפתחת כאחת הביטויים של התהליך האטרופי-ניווני המערכתי עם נגע עיקרי של המערכת הסטריאטלית של המוח ושל גרעין תת-גרעיני אחר. בירושה דומיננטית אוטוסומלית
ככלל, המחלה מתבטאת בעשור השלישי או הרביעי של החיים עם היפרקינזיס מקוריפורמי (בעיקר בפנים, בזרועות, בכתפיים, בהליכה), בשינויים באישיות (טיפוסים היסטריים, היסטריים וסכיזואידים של אנומליות אישיות), הפרעות פסיכוטיות (דיכאון מסוים עם קדרות, קדרות, דיספוריה; מצב רוח פרנואידי).
חשיבות מיוחדת לאבחון היא שילוב של היפרקינזיס של כוריופורם, דמנציה ונטל תורשתי. להלן ספציפיות דמנציה זו:
- (10-15 שנים בממוצע): דיסוציאציה בין היכולת המתמשכת לדון בעצמך לבין חוסר עקביות אינטלקטואלי לכאורה במצבים הדורשים עבודה נפשית פרודוקטיבית (חשיבה מושגית, לימוד דברים חדשים);
- אי-סדירות חריגה של תפקוד מנטלי, המבוססת על הפרות חמורות של תשומת לב ושל חוסר עקביות של עמדות המטופל (חשיבה "פתאומית", על ידי אנלוגיה עם היפרקינזיס);
- חוסר תוחלת של הפרות ברורות של תפקודים קליפתיים גבוהים;
- הקשר ההפוך בין הגידול בדמנציה לבין חומרת ההפרעות הפסיכוטיות.
אם ניקח בחשבון את שיעור גבוה של אשליות פרנואידית של קנאה, רדיפה) והפרעות דיספוריות בתמונה הקלינית של המחלה, הטיפול מתבצע באמצעות נוירופלטיקה שונים החוסמים קולטנים דופאמינרגיים (פנוטיאזין ו butyrophhenone נגזרים) או להפחית את רמת הדופמין ברקמות (reserpine).
ההלופרידול (2-20 מ"ג ליום), tiaprid (100-600 מ"ג ליום) למשך לא יותר משלושה חודשים, thioridazine (עד 100 מ"ג ליום), reserpine (0.25-2 מ"ג / יום), נוגדת פרכוסים clonazepam (1, 5-6 מ"ג ליום). תרופות אלה תורמות להפחתת היפרקינציה, החלקה של מתח רגשי, פיצוי הפרעות אישיות.
בבית החולים, הטיפול בהפרעות נפשיות מתבצע תוך התחשבות בתסמונת המובילה, גיל ומצבו הכללי של המטופל. בטיפול החוץ, עקרונות הטיפול הם זהים (טיפול תחזוקה מתמשך של הפרעות תנועה, החלפה תקופתית של התרופה). טיפול חוץ במינונים נמוכים יותר של נוירופלטיקה.
פעילויות שיקום עבור דמנציה קלה עד בינונית כוללות טיפול תעסוקתי, פסיכותרפיה, והכשרה קוגניטיבית. יש צורך לעבוד עם בני משפחה, תמיכה פסיכולוגית של אנשים המטפלים בחולים. השיטה העיקרית למניעת המחלה היא ייעוץ רפואי וגנטי של קרובי משפחתו של המטופל עם הפניה לניתוח DNA בבואו להחליט אם ללדת.
הפרוגנוזה היא בדרך כלל שלילית. מהלך המחלה מתקדם לאט, המחלה מובילה בדרך כלל למוות תוך 10-15 שנים.
מה מטריד אותך?
טיפול במחלת הנטינגטון
הטיפול במחלת הנטינגטון הוא סימפטומטי. Chorea וחרדה ניתן לדכא חלקית על ידי neuroleptics (למשל, chlorpromazine 25-300 מ"ג בעל פה 3 פעמים ביום, haloperidol 5-45 מ"ג בעל פה 2 פעמים ביום) או reserpine 0.1 מ"ג דרך הפה 1 שעה / יום. מינון מוגבר עד למקסימום נסבל (עד תופעות לוואי להופיע, כגון נמנום, פרקינסון, עבור reserpine, לחץ דם). מטרת הטיפול האמפירי היא להפחית את התמסורת הגלוטמטרגית באמצעות קולטני Nmethyl-O-aspartate ולתמוך בייצור אנרגיה במיטוכונדריה. טיפול שמטרתו הגדלת GABA במוח אינו יעיל.
בדיקה גנטית וייעוץ חשובים משום שהתסמינים של המחלה באים לידי ביטוי בסוף גיל הפוריות. אנשים עם היסטוריה משפחתית חיובית ומעוניינים בבדיקה נשלחים למרכזים מיוחדים, תוך התחשבות בכל ההשלכות האתיות והפסיכולוגיות.
טיפול סימפטומטי במחלת הנטינגטון
טיפול יעיל שיכול לעצור את התקדמות המחלה של הנטינגטון עדיין לא פותח. בדיקות שנערכו שוב ושוב של תרופות שונות, אבל כדי להשיג כל השפעה משמעותית לא היה אפשרי. נוירופלטיקה ועוד אנטגוניסטים של קולטן דופאמין נמצאים בשימוש נרחב לתיקון הפרעות נפשיות ותנועות לא רצוניות בחולים עם מחלת הנטינגטון. תנועות בלתי רצוניות משקפות חוסר איזון בין מערכות דופאמינרגיות ו GABAergic. בהתאם לכך, תרופות אנטי-פסיכוטיות משמשות להפחתת הפעילות הדופאמינרגית העודפת. עם זאת, תרופות אלה עצמם יכולים לגרום בולט תופעות לוואי קוגניטיבית ו extrapyramidal. יתר על כן, למעט מקרים שבהם המטופל מפתח פסיכוזה או עוררות, יעילותם לא הוכחה. Neuroleptics לעתים קרובות לגרום או להחריף dysphagia או הפרעות תנועה אחרות. Neuroleptics של הדור החדש, כגון risperidone, clozapine ו olanzapine, עשוי להיות שימושי במיוחד לטיפול במחלת הנטינגטון, שכן הם גורמים תופעות לוואי extrapyramidal במידה פחותה, אבל יכול להחליש תסמונת פרנואידית או עצבנות מוגברת.
Tetrabenazine ו reserpine גם להחליש את הפעילות של המערכת דופאמינרגי יכול להפחית את חומרת תנועות לא רצוניות בשלב מוקדם של המחלה. עם זאת, תרופות אלה יכולים לגרום לדיכאון. מאז המחלה עצמה לעיתים קרובות גורם לדיכאון, תופעת לוואי זו מגבילה באופן משמעותי את השימוש reserpine ו tetrabenazine. בשלב מאוחר של המחלה, התאים הנושאת קולטני דופאמין מתים, ולכן האפקטיביות של אנטגוניסטים של קולטן דופאמין נחלשת או הולכת לאיבוד.
נוירופלטיקה, תרופות נוגדות דיכאון וחרדות משמשות לטיפול בפסיכוזה, דיכאון ועצבנות בחולים עם מחלת הנטינגטון, אך יש לרשום אותם רק לתקופה בה החולה סובל מתסמינים אלו. תרופות אשר עשויות להיות שימושיות בשלב אחד של המחלה, כפי שהיא מתקדמת, עשוי להיות יעיל או אפילו השפעה שלילית.
בחולים עם מחלת הנטינגטון נבדקו אגוניסטים של קולטן GABA, משום שחולי הנטינגטון חשפו ירידה משמעותית ברמות GABA בסטריאטום, וכן רגישות יתר של קולטני GABA באזורי ההקרנה. בנזודיאזפינים הוכיחו יעילות במקרים בהם תנועות לא רצונית וקשיי קוגניטיבית מחריפים על ידי מתח וחרדה. מינונים נמוכים של תרופות אלה צריכים להיות prescribed כדי למנוע הרגעה לא רצויה. ברוב החולים עם מחלת הנטינגטון, אף אחת מהתרופות לא מובילה לשיפור משמעותי באיכות החיים.
עם תחילתו המוקדמת של מחלת הנטינגטון, המתרחשת בסימפטומים של פרקינסון, ניתן לנסות את הדופאמינרגים, אך יעילותם מוגבלת. יתר על כן, levodopa יכול לגרום או לחזק מיוקלונוס בחולים אלה. יחד עם זאת, בקלופן יכול להפחית את הנוקשות אצל חלק מהחולים במחלת הנטינגטון.
טיפול מונע (נוירופרוטטיבי) במחלת הנטינגטון
למרות המום הגנטי של הנטינגטון של המחלה ידוע, עדיין לא ברור איך זה מוביל ניוון סלקטיבי של נוירונים. הוא האמין כי טיפול מונע שמטרתו להפחית את הלחץ החמצוני ואת ההשפעה excitotoxic הוא מסוגל מסוגל להאט או להשעות את התקדמות המחלה. המצב עשוי, במובנים מסוימים, להידמות להתנוונות הכבדית, שבה המום הגנטי נשאר בלתי ידוע במשך שנים רבות, אולם טיפול מניעתי שמטרתו השפעה משנית - הצטברות של נחושת - הובילה ל"ריפוי ". בהקשר זה, ההשערה כי מחלת הנטינגטון קשורה להפרעה של מטבוליזם של אנרגיה ומוות של תאים עקב השפעה אקסיטוטוקסית, מושכת תשומת לב מיוחדת. המחלה עצמה עלולה לגרום למוות של תאים בשל הצטברות intranuclear של שברי מסוף N- של תפקודי הסלולר והמטבולי המפרקים. תהליך זה יכול להשפיע על כמה קבוצות של נוירונים במידה רבה יותר מאשר קבוצות אחרות, בשל הרגישות הגבוהה שלהם נזק excitotoxic. במקרה זה, טיפול מונע עם אנטגוניסטים של קולטן חומצות אמיניות מעורר או אמצעי למניעת נזק רדיקלים חופשיים יוכל למנוע או לעכב את ההתקדמות וההתקדמות של המחלה. במודלים מעבדה של טרשת לרוחב אמיוטרופית, הוכח כי חומרים נוגדי חמצון וקולטן אנטגוניסטים (HAC) יכול להאט את התקדמות המחלה. גישות דומות יכולות להיות יעילות במחלת הנטינגטון. נכון לעכשיו, ניסויים קליניים מתנהלים על אנטגוניסטים קולטן גלוטמט וסוכנים כי לשפר את הפונקציה של II המורכב של שרשרת המיטוכונדריה התחבורה האלקטרון.