המומחה הרפואי של המאמר

פרסומים חדשים
מחלת הנטינגטון
סקירה אחרונה: 05.07.2025

כל תוכן iLive נבדק מבחינה רפואית או נבדק למעשה כדי להבטיח דיוק עובדתי רב ככל האפשר.
יש לנו קווים מנחים קפדניים המקור רק קישור לאתרים מדיה מכובד, מוסדות מחקר אקדמי, בכל עת אפשרי, עמיתים מבחינה רפואית מחקרים. שים לב שהמספרים בסוגריים ([1], [2] וכו ') הם קישורים הניתנים ללחיצה למחקרים אלה.
אם אתה סבור שתוכן כלשהו שלנו אינו מדויק, לא עדכני או מפוקפק אחרת, בחר אותו ולחץ על Ctrl + Enter.
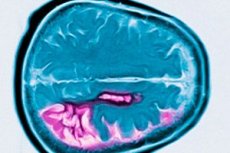
מחלת הנטינגטון היא הפרעה ניוונית עצבית אוטוזומלית דומיננטית המאופיינת בירידה קוגניטיבית מתקדמת, תנועות לא רצוניות ופגיעה בקואורדינציה מוטורית החל מגיל העמידה. האבחון מאושר על ידי בדיקה גנטית. הטיפול הוא בעיקר סימפטומטי. בדיקות גנטיות עשויות להיות מומלצות עבור קרובי משפחה. ג'ורג' הנטינגטון תיאר לראשונה את המצב בשנת 1872, לאחר שחקר מקרה משפחתי בתושבי לונג איילנד.
שכיחות מחלת הנטינגטון היא כ-10 מקרים לכל 100,000 תושבים, ובהתחשב בהופעתה המאוחרת, כ-30 איש מתוך 100,000 נמצאים בסיכון של 50% לפתח אותה במהלך חייהם. למרות שהמחלה מופיעה לרוב בין הגילאים 35 ל-40, טווח הגילאים בו היא מופיעה הוא רחב למדי, כאשר ההופעה המוקדמת ביותר היא בגיל 3 והמאוחרת ביותר בגיל 90. למרות שבתחילה נחשבה חדירה של המחלה ל-100%, כיום מאמינים שזה לא תמיד המקרה. אצל אנשים שירשו את הגן למחלה מאביהם, המחלה מתבטאת בממוצע 3 שנים מוקדם יותר מאשר אצל אלו שירשו את הגן הפתולוגי מאמם. בכ-80% מהחולים שירשו את הגן הפתולוגי מאביהם, המחלה מתבטאת לפני גיל 20. התופעה של ביטוי מוקדם יותר של פגם גנטי בצאצאים נקראת ציפייה.
 [ 1 ]
[ 1 ]
מה גורם למחלת הנטינגטון?
למחלת הנטינגטון אין העדפה מגדרית. מוצגת אטרופיה של הגרעין הזנבי, שבו נוירונים קטנים מתנוונים ורמת הנוירוטרנסמיטרים - חומצה גמא-אמינובוטירית (GABA) וחומר P - יורדת.
גן מוטנטי עם מספר מוגבר ("התרחבות") של רצפי DNA של CAG (ציסטאין-אלנין-גליצין) המקודדים לחומצת האמינו גלוטמין אחראי להתפתחות מחלת הנטינגטון. התוצר של גן זה, החלבון הגדול הנטינגטין, מכיל כמות מוגזמת של שיירי פוליגלוטמין, מה שמוביל למחלה באמצעות מנגנון לא ידוע. ככל ש-CAG חזרות יותר, כך המחלה מופיעה מוקדם יותר ומהלכה חמור יותר. מדור לדור, מספר החזרות יכול לעלות, מה שמוביל עם הזמן להחמרה של הפנוטיפ המשפחתי.
למרות עניין רב בשינויים הגנטיים והביוכימיים במחלת פרקינסון, החיפוש אחר גן למחלה לא צלח עד סוף שנות ה-70. באותה תקופה, ננסי וקסלר ואלן טובין ארגנו סדנה בחסות קרן המחלות התורשתיות כדי לדון באסטרטגיה למציאת גן למחלת הנטינגטון. דיוויד האוסמן, דיוויד בוטשטיין וריי ווייט, שהשתתפו בפגישה, הציעו כי טכניקות DNA רקומביננטי שפותחו לאחרונה עשויות לסייע בהשגת מטרה זו. משימה מרכזית בפרויקט הייתה למצוא משפחה גדולה עם דורות רבים של מחלת הנטינגטון כדי להשיג דגימות DNA. בשנת 1979 הושק פרויקט משותף של מדענים מוונצואלה ומארצות הברית לבחון משפחה גדולה עם מחלת הנטינגטון שחיה לחופי אגם מאראצ'ייבו (ונצואלה). בשנת 1983, גן מחלת הנטינגטון אותר בקצה הזרוע הקצרה של כרומוזום 4 (Gusella et al., 1983), ועשור לאחר מכן התגלה כי המוטציה של גן זה מורכבת מעלייה במספר החזרות של הטרינוקלאוטיד ציטוזין-אדנין-גואנין (CAG) (Huntington's Disease Collaborative Research Group, 1993). המתודולוגיה שפותחה על ידי קבוצה מדעית זו נחשבת כיום לסטנדרטית לשיבוט מיקומי של גנים חדשים.
בעוד שלגן הבר יש טווח של 10-28 חזרות CAG, לצורה המוטנטית של הגן הגורם למחלת הנטינגטון יש טווח מוגבר מ-39 ליותר מ-100 חזרות CAG. גילוי הרחבת החזרות הטרינוקלאוטידיות סייע להסביר רבים מהמאפיינים הקליניים של המחלה. בפרט, נמצא מתאם הפוך בין גיל הופעת המחלה לאורך האזור עם טרינוקלאוטידים חוזרים. ניתן להסביר את הציפייה לתורשה אבהית על ידי העובדה שעלייה במספר החזרות מתרחשת לעיתים קרובות אצל גברים במהלך יצירת זרע. ניתוח מוטציות חדשות הראה שהן מתרחשות בדרך כלל כאשר לאחד ההורים, בדרך כלל האב, הייתה ספירת חזרות CAG גבוהה מ-28; במקרה זה, מספר החזרות הללו גדל בדור הבא. כעת נקבע שאם מספר החזרות אינו עולה על 28, הוא מועבר ביציבות מדור לדור. אם מספר החזרות הוא בין 29 ל-35, אז תסמיני מחלת הנטינגטון אינם מופיעים, אך כאשר היא מועברת לצאצאים, אורך אזור זה עשוי לגדול. אם מספר החזרות הוא בין 36 ל-39, אז במקרים מסוימים (אך לא תמיד) המחלה עשויה להתבטא קלינית (חדירה לא שלמה), וכאשר היא מועברת לצאצאים, עלייה במספר החזרות הטרינוקלאוטידיות אפשרית. אם מספר החזרות עולה על 40, אז המחלה מופיעה כמעט בכל המקרים, וכאשר היא מועברת לצאצאים, התרחבות נוספת של החזרות אפשרית. הסיבות לעלייה במספר החזרות נותרות לא ידועות.
פתומורפולוגיה של מחלת הנטינגטון
מחלת הנטינגטון מאופיינת באובדן נוירונים בעיקר בגרעין הזנב ובפוטמן, ובמידה מסוימת גם בקליפת המוח ובמבנים אחרים במוח. משקל המוח הכולל במחלת הנטינגטון מצטמצם לא רק עקב ירידה במספר הנוירונים, אלא גם עקב אובדן של חומר לבן. בקליפת המוח, תאים בשכבות V ו-VI מושפעים ביותר. חומרת השינויים הניווניים המיקרו-מקרוסקופיים והמקרוסקופיים (מותאמים לגיל בעת המוות) מתואמת עם מספר חזרות CAG. ניתוח פתולוגי מפורט של שינויים בכמה מאות מקרים של מחלת הנטינגטון הראה כי ניוון הסטריאטום מתחיל בחלק הדורסו-מדיאלי של גרעין הזנב ובחלק הדורסולטרלי של הפוטמן, ולאחר מכן מתפשט לגחוני. קבוצות שונות של נוירונים בגרעין הזנב ובפוטמן מושפעות בדרגות שונות. נוירונים פנימיים בסטריאטום נשארים שלמים יחסית, אך חלק מהנוירונים ההקרנתיים מושפעים באופן סלקטיבי. בצורה הצעירה של מחלת הנטינגטון, שינויים פתומורפולוגיים בסטריאטום בולטים ונרחבים יותר, וכוללים את קליפת המוח, המוח הקטן, התלמוס והגלובוס פלידוס.
שינויים נוירוכימיים במחלת הנטינגטון
GABA. מחקרים נוירוכימיים של המוח בחולים עם מחלת הנטינגטון גילו ירידה משמעותית בריכוז GABA בסטריאטום. מחקרים מאוחרים יותר אישרו כי מחלת הנטינגטון קשורה לירידה במספר הנוירונים ה-GABA-ארגיים והראו כי ריכוזי GABA מופחתים לא רק בסטריאטום אלא גם באזורי ההקרנה שלו - המקטעים החיצוניים והפנימיים של הגלובוס פלידוס והסובסטנציה ניגרה. במוח במחלת הנטינגטון, שינויים בקולטני GABA זוהו גם באמצעות מחקרי קישור קולטנים והכלאה in situ של mRNA. מספר קולטני ה-GABA הופחת במידה בינונית בגרעין הזנב ובפוטמן, אך גדל בחלק הרטיקולרי של הסובסטנציה ניגרה ובמקטע החיצוני של הגלובוס פלידוס, ככל הנראה עקב רגישות יתר לדה-עצבוב.
אצטילכולין. אצטילכולין משמש כמוליך עצבי על ידי נוירונים פנימיים גדולים שאינם קוצניים בסטריאטום. מחקרים מוקדמים שלאחר המוות בחולים עם מחלת הנטינגטון הראו ירידה בפעילות כולין אצטילטרנספראז (ChAT) בסטריאטום, דבר המצביע על אובדן של נוירונים כולינרגיים. עם זאת, בהשוואה לירידה המשמעותית בנוירונים גאבא-ארגיים, נוירונים פנימיים כולינרגיים חסכים יחסית. לכן, צפיפות הנוירונים החיוביים לאצטילכולין אסטראז ופעילות ChAT בסטריאטום גבוהות יחסית בהשוואה לקבוצת ביקורת תואמת גיל.
חומר P. חומר P נמצא בנוירונים קוצניים בינוניים רבים בסטריאטום, אשר מוקרנים בעיקר אל המקטע הפנימי של הגלובוס פלידוס והסובסטנטיה ניגרה, ובדרך כלל מכילים גם דינורפין ו-GABA. רמות חומר P בסטריאטום ובחלק הרטיקולרי של הסובסטנטיה ניגרה מופחתות במחלת הנטינגטון. בשלב הסופי של המחלה, מחקרים אימונוהיסטוכימיים גילו ירידה משמעותית במספר הנוירונים המכילים חומר P. בשלבים מוקדמים יותר, נוירונים המכילים חומר P ובולטים אל המקטע הפנימי של הגלובוס פלידוס חסכים יחסית, בהשוואה לנוירונים הבולטים אל החלק הרטיקולרי של הסובסטנטיה ניגרה.
פפטידים אופיואידים. אנקפלין נמצא בנוירונים גאבא-ארגיים בעלי התקדמות קוצנית בינונית של המסלול העקיף, אשר מוקרנים אל המקטע החיצוני של הגלובוס פלידוס ונושאים קולטני D2. מחקרים אימונוהיסטוכימיים הראו כי נוירונים המכילים אנקפלין המוקרנים אל המקטע החיצוני של הגלובוס פלידוס אובדים בשלב מוקדם של מחלת הנטינגטון. תאים אלה מתים ככל הנראה מוקדם יותר מתאים המכילים חומר P המוקרנים אל המקטע הפנימי של הגלובוס פלידוס.
קטכולאמינים. נוירונים המכילים אמינים ביוגניים (דופמין, סרוטונין) ובולטים אל הסטריאטום ממוקמים בחלק הקומפקטי של הסובסטנציה ניגרה, הטגמנטום הגחוני וגרעיני הרפה. בעוד שהבליטות הנוראדרנרגיות אל הסטריאטום האנושי הן מינימליות, רמות הסרוטונין והדופמין (לגרם של רקמה) בסטריאטום גבוהות, דבר המצביע על שימור הבליטות האפרנטיות הללו למרות האובדן המשמעותי של הנוירונים של הסטריאטום עצמו. נוירונים דופמינרגיים של הסובסטנציה ניגרה נותרים שלמים הן בצורות הקלאסיות והן בצורות הצעירות של מחלת הנטינגטון.
סומטוסטטין/נוירופפטיד Y וסינתטאז תחמוצת החנקן. מדידת רמות סומטוסטטין ונוירופפטיד Y בסטריאטום במחלת הנטינגטון גילתה עלייה פי 4-5 בהשוואה לרקמות תקינות. מחקרים אימונוהיסטוכימיים הראו שימור מוחלט של נוירונים פנימיים בסטריאטום המכילים נוירופפטיד Y, סומטוסטטין וסינתטאז תחמוצת החנקן. לפיכך, נוירונים אלה עמידים לתהליך הפתולוגי.
חומצות אמינו מעוררות. הוצע כי מוות תאי סלקטיבי במחלת הנטינגטון נובע מהשפעה נוירוטוקסית הנגרמת על ידי גלוטמט. רמות הגלוטמט וחומצה כינולינית (נוירוטוקסין אנדוגני שהוא תוצר לוואי של מטבוליזם של סרוטונין ואגוניסט של קולטני גלוטמט) בסטריאטום של מחלת הנטינגטון משתנות מעט, אך מחקר שנערך לאחרונה באמצעות ספקטרוסקופיית MR גילה עלייה ברמות הגלוטמט in vivo. רמת האנזים הגליאלי האחראי לסינתזה של חומצה כינולינית בסטריאטום של מחלת הנטינגטון עולה פי 5 בערך בהשוואה לנורמה, בעוד שפעילות האנזים המבטיח את פירוק החומצה הכינולינית עולה במחלת הנטינגטון רק ב-20-50%. לפיכך, הסינתזה של חומצה כינולינית עשויה להיות מוגברת במחלת הנטינגטון.
מחקרים על קולטני חומצות אמינו מעוררות (EAA) במחלת הנטינגטון גילו ירידה משמעותית במספר קולטני NMDA, AMPA, קיינט וגלוטמט מטאבוטרופי בסטריאטום, כמו גם קולטני AMPA וקיינט בקליפת המוח. בשלב המאוחר של מחלת הנטינגטון, קולטני NMDA כמעט ולא היו קיימים, בעוד שבשלבים הפרה-קליניים והמוקדמים נצפתה ירידה משמעותית במספר קולטנים אלה.
רגישות סלקטיבית. במחלת הנטינגטון, סוגים מסוימים של תאים סטריאטליים אובדים באופן סלקטיבי. הנוירונים הקוצניים הבינוניים, אשר מביטים אל החלק החיצוני של הגלובוס פלידוס ומכילים גאבא ואנקפלין, מתים בשלב מוקדם מאוד של המחלה, וכך גם הנוירונים המכילים גאבא וסובסטנס P ומביטים אל החלק הרטיקולרי של הגלובוס ניגרה. אובדן הנוירונים המכילים גאבא ואנקפלין ומביטים אל החלק החיצוני של הגלובוס ניגרה מבטל את העיכוב של מבנה זה, מה שמוביל בתורו לעיכוב פעיל של הגרעין התת-תלמוסי. הירידה בפעילות של הגרעין התת-תלמוסי יכולה ככל הנראה להסביר את התנועות הכוריאוגרפיות המתרחשות במחלת הנטינגטון. ידוע זה מכבר כי נגעים מוקדיים של הגרעין התת-תלמוסי יכולים לגרום לכוריאה. אובדן של נוירונים מסוג גאבא וסובסטנס P המביטים אל הגלובוס ניגרה חלק רטיקולריס עשוי להיות אחראי להפרעות האוקולומוטוריות הנראות במחלת הנטינגטון. מסלול זה בדרך כלל מעכב נוירונים מסוג substantia nigra pars reticularis המוקרנים אל ה-superior colliculus, אשר בתורם מווסתים את הסקאדות. במחלת הנטינגטון לנוער, המסלולים שהוזכרו לעיל מושפעים בצורה חמורה יותר, ובנוסף, בליטות סטריאטליות אל המקטע הפנימי של ה-globus pallidus אובדות מוקדם.
חלבון הנטינגטין, המקודד על ידי הגן שהמוטציה שלו גורמת למחלת הנטינגטון, נמצא במבנים שונים של המוח וברקמות אחרות. הנטינגטין נמצא בדרך כלל בעיקר בציטופלזמה של נוירונים. החלבון נמצא ברוב הנוירונים במוח, אך נתונים עדכניים מראים שתכולתו גבוהה יותר בנוירוני מטריקס מאשר בנוירונים סטריוזומליים, וגבוה יותר בנוירוני השלכה מאשר בנוירונים פנימיים. לפיכך, הרגישות הסלקטיבית של נוירונים מתואמת עם תכולת הנטינגטין שלהם, אשר בדרך כלל קיימת באוכלוסיות נוירונים מסוימות.
כמו במוחם של חולי מחלת הנטינגטון, בעכברים טרנסגניים עבור הפרגמנט ה-N-טרמינלי של גן מחלת הנטינגטון עם מספר מורחב של חזרות, הנטינגטין יוצר אגרגטים צפופים בגרעיני הנוירונים. תכלילים תוך-גרעיניים אלה נוצרים בנוירונים של השלכה סטריאטלית (אך לא בנוירונים פנימיים). בעכברים טרנסגניים, התכלילים נוצרים מספר שבועות לפני הופעת התסמינים. נתונים אלה מצביעים על כך שחלבון הנטינגטין המכיל מספר מוגבר של שיירי גלוטמין שתכליליהם מקודדים חזרות טרינוקלאוטיד, או פרגמנט שלו, מצטבר בגרעין התא וכתוצאה מכך עלול לפגוע בשליטתו על תפקודים תאיים.
תסמינים של מחלת הנטינגטון
קשה לקבוע במדויק את הגיל בו הופיעו התסמינים הראשונים אצל חולים במחלת הנטינגטון, שכן המחלה מתבטאת בהדרגה. שינויים באישיות ובהתנהגות, הפרעות קואורדינציה קלות, עשויים להתרחש שנים רבות לפני הופעת תסמינים ברורים יותר. עד לקביעת האבחון, לרוב החולים יש תנועות כוריאולוגיות, קואורדינציה לקויה של תנועות עדינות ויצירת תנועות תנועתיות רצוניות איטית. ככל שהמחלה מתקדמת, היכולת לארגן את הפעילויות נפגעת, הזיכרון יורד, הדיבור מתקשה, הפרעות אוקולומוטוריות וביצועים לקויים של תנועות מתואמות גוברים. למרות שבשלב המוקדם של המחלה אין שינויים בשרירים וביציבה, עם התקדמותה עלולות להתפתח תנוחות דיסטוניות, אשר עם הזמן עלולות להפוך לתסמין דומיננטי. בשלב מאוחר, הדיבור הופך לבלתי צפוי, הבליעה מתקשה משמעותית, ההליכה הופכת לבלתי אפשרית. מחלת הנטינגטון מתקדמת בדרך כלל במשך 15-20 שנים. בשלב הסופי, החולה חסר אונים ודורש טיפול מתמיד. התוצאה הקטלנית אינה קשורה ישירות למחלה הראשונית, אלא לסיבוכיה, למשל, דלקת ריאות.
דמנציה במחלת הנטינגטון
קוד ICD-10
P02.2. דמנציה במחלת הנטינגטון (G10).
דמנציה מתפתחת כאחת הביטויים של תהליך ניווני-אטרופי מערכתי עם נזק דומיננטי למערכת הסטריאטלית של המוח ולגרעינים תת-קוקליים אחרים. היא עוברת בתורשה באופן אוטוזומלי דומיננטי.
ככלל, המחלה מתבטאת בעשור השלישי או הרביעי לחיים עם היפרקינזיס כוריאוגרפית (במיוחד בפנים, בזרועות, בכתפיים, בהליכה), שינויים באישיות (אנומליות אישיות מסוגים נרגשים, היסטריים וסכיזואידיים), הפרעות פסיכוטיות (דיכאון מיוחד עם קדרות, זעף, דיספוריה; מצב רוח פרנואידי).
חשיבות מיוחדת לאבחון היא השילוב של היפרקינזיס כוריאוגרפית, דמנציה ועומס תורשתי. להלן הספציפיים לדמנציה זו:
- התקדמות איטית (בממוצע 10-15 שנים): ניתוק בין היכולת שנותרה לדאוג לעצמך לבין חוסר יכולת אינטלקטואלית ברור במצבים הדורשים עבודה מנטלית פרודוקטיבית (חשיבה מושגית, לימוד דברים חדשים);
- חוסר אחידות בולט של ביצועים נפשיים, המבוסס על הפרעות קשב גסות וחוסר עקביות בגישותיו של המטופל ("חשיבה קופצנית", בדומה להיפרקינזיס);
- חריגות של הפרות ברורות של תפקודים קורטיקליים גבוהים יותר;
- קשר הפוך בין העלייה בדמנציה לבין חומרת ההפרעות הפסיכוטיות.
בהתחשב בשיעור הגבוה של הפרעות פסיכוטיות (הזיות פרנואידיות של קנאה, רדיפה) והפרעות דיספוריות בתמונה הקלינית של המחלה, הטיפול מתבצע באמצעות תרופות נוירולפטיות שונות החוסמות קולטנים דופמינרגיים (נגזרות פנוטיאזין ובוטירופנון) או מפחיתות את רמת הדופמין ברקמות (רסרפין).
משתמשים בהלופרידול (2-20 מ"ג/יום), טיאפריד (100-600 מ"ג/יום) למשך לא יותר משלושה חודשים, תיאורידזין (עד 100 מ"ג/יום), רסרפין (0.25-2 מ"ג/יום) והתרופה נוגדת הפרכוסים קלונאזפאם (1.5-6 מ"ג/יום). תרופות אלו מסייעות בהפחתת היפרקינזיס, החלשת מתח רגשי ופיצוי על הפרעות אישיות.
טיפול באשפוז בהפרעות נפשיות מתבצע תוך התחשבות בתסמונת המובילה, בגיל ובמצב הכללי של המטופל. בטיפול אמבולטורי, עקרונות הטיפול זהים (טיפול תחזוקה מתמשך בהפרעות תנועה, החלפה תקופתית של תרופה). בטיפול אמבולטורי משתמשים במינונים נמוכים יותר של תרופות נוירולפטיות.
אמצעי שיקום לדמנציה קלה ובינונית כוללים ריפוי בעיסוק, פסיכותרפיה ואימון קוגניטיבי. יש צורך לעבוד עם בני משפחה ולספק תמיכה פסיכולוגית לאנשים המטפלים בחולה. השיטה העיקרית למניעת מחלות היא ייעוץ רפואי וגנטי לקרובי משפחתו הקרובים ביותר של החולה עם הפניה לניתוח DNA בעת קבלת החלטה על לידה.
הפרוגנוזה בדרך כלל שלילית. מהלך המחלה מתקדם באיטיות, והמחלה מובילה בדרך כלל למוות לאחר 10-15 שנים.
[ 18 ]
מה מטריד אותך?
טיפול במחלת הנטינגטון
הטיפול במחלת הנטינגטון הוא סימפטומטי. ניתן לדכא חלקית את הכוריאה והתסיסה באמצעות תרופות נוירולפטיות (למשל, כלורפרומזין 25-300 מ"ג דרך הפה 3 פעמים ביום, הלופרידול 5-45 מ"ג דרך הפה פעמיים ביום) או רסרפין 0.1 מ"ג דרך הפה פעם ביום. יש להעלות את המינונים למקסימום הנסבל (לפני הופעת תופעות לוואי, כגון נמנום, פרקינסון; עבור רסרפין, לחץ דם נמוך). מטרת הטיפול האמפירי היא להפחית את ההעברה הגלוטמטרגית דרך קולטני Nmethyl-O-aspartate ולשמור על ייצור אנרגיה במיטוכונדריה. טיפול שמטרתו להגביר את GABA במוח אינו יעיל.
בדיקות גנטיות וייעוץ חשובים משום שתסמיני המחלה מופיעים לאחר שנות הפוריות. אנשים עם היסטוריה משפחתית חיובית ואלו המעוניינים בבדיקות מופנים למרכזים ייעודיים, תוך התחשבות בכל ההשלכות האתיות והפסיכולוגיות.
טיפול סימפטומטי במחלת הנטינגטון
אין טיפול יעיל שיכול לעצור את התקדמות מחלת הנטינגטון. נערכו מספר ניסויים בתרופות שונות, אך לא הושגה השפעה משמעותית. נוירולפטיקה ואנטגוניסטים אחרים של קולטני דופמין נמצאים בשימוש נרחב לתיקון הפרעות נפשיות ותנועות לא רצוניות בחולים עם מחלת הנטינגטון. תנועות לא רצוניות משקפות חוסר איזון בין המערכות הדופמינרגיות וה-GABA-ארגיות. בהתאם, נוירולפטיקה משמשת להפחתת פעילות דופמינרגית עודפת. עם זאת, תרופות אלו עצמן עלולות לגרום לתופעות לוואי קוגניטיביות ואקסטראפירמידליות משמעותיות. בנוסף, למעט במקרים בהם המטופל מפתח פסיכוזה או תסיסה, יעילותן לא הוכחה. נוירולפטיקה גורמת או מחמירה לעיתים קרובות דיספאגיה או הפרעות תנועה אחרות. נוירולפטיקה מדור חדש יותר כמו ריספרידון, קלוזפין ואולנזפין עשויה להיות שימושית במיוחד בטיפול במחלת הנטינגטון מכיוון שהיא גורמת לפחות תופעות לוואי אקסטראפירמידליות אך עשויה להפחית תסמינים פרנואידיים או עצבנות מוגברת.
טטרבנזין ורסרפין גם מפחיתים את פעילות המערכת הדופמינרגית ויכולים להפחית את חומרת התנועות הלא רצוניות בשלבים המוקדמים של המחלה. עם זאת, תרופות אלו עלולות לגרום לדיכאון. מכיוון שהמחלה עצמה גורמת לעיתים קרובות לדיכאון, תופעת לוואי זו מגבילה משמעותית את השימוש ברסרפין וטטרבנזין. בשלבים המאוחרים של המחלה, תאים הנושאים קולטני דופמין מתים, כך שהיעילות של אנטגוניסטים לקולטני דופמין נחלשת או אובדת.
נוירולפטיקה, תרופות נוגדות דיכאון וחרדה משמשות לטיפול בפסיכוזה, דיכאון ועצבנות אצל חולים במחלת הנטינגטון, אך יש לרשום אותן רק כל עוד לחולה יש תסמינים אלה בפועל. תרופות שעשויות להיות מועילות בשלב מסוים של המחלה עלולות להפוך ללא יעילות או אפילו מזיקות ככל שהמחלה מתקדמת.
אגוניסטים של קולטני GABA נבדקו בחולים עם מחלת הנטינגטון, מכיוון שנמצא כי במחלת הנטינגטון יש ירידה משמעותית ברמות GABA בסטריאטום, כמו גם רגישות יתר של קולטני GABA באזורי ההקרנה שלה. בנזודיאזפינים הוכחו כיעילים במקרים בהם תנועות לא רצוניות ופגיעה קוגניטיבית מחמירות עקב לחץ וחרדה. יש לרשום מינונים נמוכים של תרופות אלו כדי להימנע מהשפעות הרגעה לא רצויות. ברוב החולים עם מחלת הנטינגטון, אף אחת מהתרופות לא מובילה לשיפור משמעותי באיכות החיים.
במחלת הנטינגטון מוקדמת עם תסמיני פרקינסון, ניתן לנסות תרופות דופמינרגיות, אך יעילותן מוגבלת. יתר על כן, לבודופה עלולה לגרום או להגביר מיוקלונוס בחולים אלו. במקביל, בקלופן עשוי להפחית נוקשות אצל חלק מהחולים במחלת הנטינגטון.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
טיפול מונע (נוירו-פרוטקטיבי) במחלת הנטינגטון
למרות שהפגם הגנטי במחלת הנטינגטון ידוע, עדיין לא ברור כיצד הוא מוביל לניוון נוירונים סלקטיבי. ההשערה היא שטיפולים מונעים שמטרתם להפחית עקה חמצונית ורעילות מעוררת עשויים להאט או לעצור את התקדמות המחלה. המצב עשוי להיות דומה במידה מסוימת לניוון הפטולנטיקולרי, שבו הפגם הגנטי נותר לא ידוע במשך שנים רבות, אך טיפולים מונעים שמטרתם להשפעה המשנית, הצטברות נחושת, "נרפאו". בהקשר זה, ההשערה שמחלת הנטינגטון קשורה להפרעה בחילוף החומרים האנרגטי ולמוות תאי עקב רעילות מעוררת משכה תשומת לב מיוחדת. המחלה עצמה עלולה לגרום למוות תאי עקב צבירה תוך-גרעינית של שברי הנטינגטין בקצה ה-N, אשר משבשת תפקודים תאיים ומטבוליים. תהליך זה עשוי להשפיע על קבוצות נוירונים מסוימות במידה רבה יותר מאחרות עקב רגישותן הגבוהה יותר לנזק מעורר. במקרה זה, טיפול מונע עם אנטגוניסטים לקולטני חומצות אמינו מעוררות או חומרים המונעים נזק מרדיקלים חופשיים יוכל למנוע או לעכב את הופעת המחלה והתקדמותה. במודלים מעבדתיים של טרשת אמיוטרופית צידית, הוכח כי חומרים נוגדי חמצון ואנטגוניסטים לקולטן (RAAs) מסוגלים להאט את התקדמות המחלה. גישות דומות עשויות להיות יעילות במחלת הנטינגטון. ניסויים קליניים של אנטגוניסטים לקולטן גלוטמט וחומרים המשפרים את תפקוד קומפלקס II של שרשרת הובלת האלקטרונים המיטוכונדריאלית נמצאים כעת בעיצומם.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]